Cell type 찾기 시리즈 : Human Cell Landscape
Celltype Annotation
Single-cell data분석에서 가장 먼저 & 기본적으로 진행하는 분석.
- 첫번째로 시도 하는 방법
- 있을법한 cell type들의 known marker gene expression을 확인해본다.
Feature Plot 예쁘게 그리는 법 - ⛔단점
- 내 생각대로 안나올 가능성 높음.
단편적으로 유전자 한두개의 발현으로 cell type결정하는 것에 대한 찝찝함.
Manual curation이라 기준이 주관적 + 시간오래 걸림 + 귀찮음
이런 단점을 보완하기 위해 다양한 cell tye annotator가 나오고 있는 추세임.
지난번에는 singleR 사용해서 reference와 비슷한 cell type을 찾는 방법을 알아봤다.
이렇게 reference data를 사용해서 cell type을 찾는 방법들의 정확도를 결정하는 요소는 바로 ‘reference가 얼마나 정확하게, 얼마나 많은 종류의 cell type을, 얼마나 자세하게 분류해서 포함하고 있는가’ 에 따라 결정된다.
이번에는 Human cell landscape라는 데이터베이스/프로젝트를 소개하고 함께 개발된 ‘single-cell HCL analysis’ pipeline을 사용해서 cell type annotation해보려고 함.
Han, X., Zhou, Z., Fei, L. et al. Construction of a human cell landscape at single-cell level. Nature 581, 303–309 (2020).
paper download
Intro
Installation
1
2
3
4
install.packages('devtools')
library(devtools)
# scHCL requires ggplot2/reshape2/plotly/shiny/shinythemes/shiny
install_github("ggjlab/scHCL")
How to use
1
2
3
4
5
6
7
8
9
10
11
12
13
library(scHCL)
# 내 데이터, seurat obj 이름 = NP
# > NP
# An object of class Seurat
# 9659 features across 169 samples within 1 assay
# Active assay: RNA (9659 features, 0 variable features)
expMat <- GetAssayData(NP, assay="RNA", slot="counts")
# scHCL has two parameters , single cell expression matrix(scdata) and
# the number of most similar cell types
hcl_result <- scHCL(scdata=expMat, numbers_plot=3)
- Run time 1 core 기준
- 200 cells -> 5 sec
~1000 cells -> 1 min
~4000 cells -> 3min
Result
1
2
3
4
5
# 결과 보기
scHCL_vis(hcl_result)
## local로 돌려서 이렇게 나옴, 서버에 설정해 놓은 IP가 있다면 그쪽으로 나올듯
#Listening on https://127.0.0.1:6234
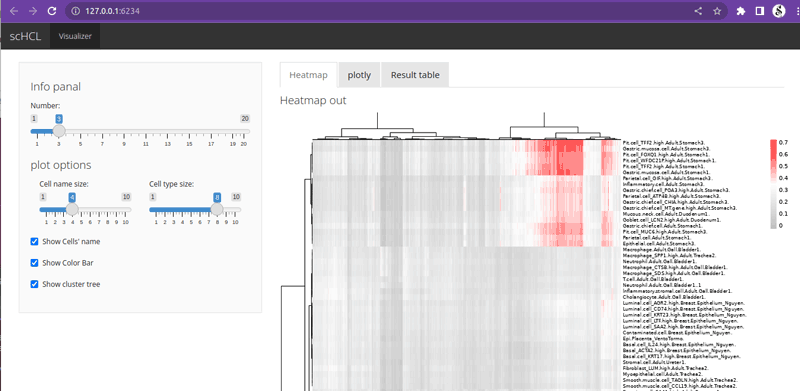 결과 확인
결과 확인
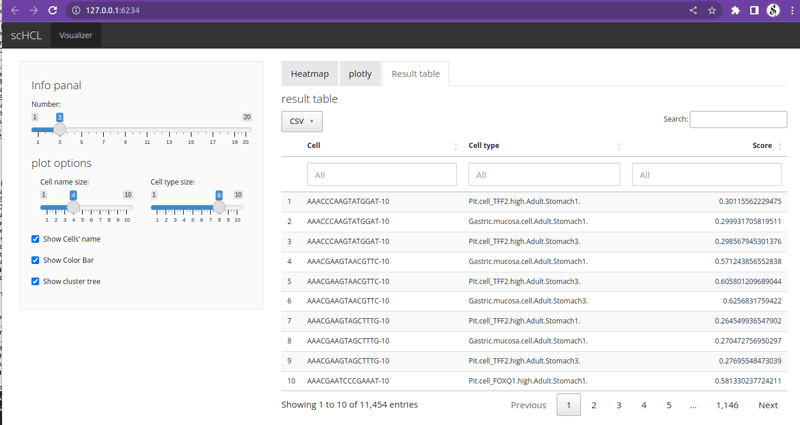 Results table tab
Results table tab
Result table 탭에서 csv 버튼 누르면 csv형식으로 된 결과 파일을 다운받을 수 있다.
R로 불러와서 👉CSV 파일 불러오기/쓰기 in R
cell type lable달아서 👉seurat cell lable달기
확인하면 됨!
Socre 가장 높은 cell type으로 label 달기
👉seurat cell lable달기 변형 버전
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
setwd("/st05/subin/Project13_PRIME/03_Seurat/PRIME06/Mark07")
# Load data
P02.TtsMepiNP <- readRDS("P02.TtsMepiNP_noIntegrat.rds") # 원래 seurat obj
scHCL_results <- read.csv("P02_scHCL.csv") # scHCL 결과
# Creat new label
## cellID별로 Score가 가장 높은 cell type 하나만 가져와서 label로 저장
P02.TtsMepiNP$scHCL_label <- sapply(colnames(P02.TtsMepiNP), function(x) as.character(subset(scHCL_results[scHCL_results$Cell==x,c("Cell.type","Score")],scHCL_results[scHCL_results$Cell==x,c("Cell.type","Score")]$Score==max(scHCL_results[scHCL_results$Cell==x,"Score"]), select=Cell.type)))
P02.TtsMepiNP$scHCL_label <- factor(P02.TtsMepiNP$scHCL_label, levels=unique(P02.TtsMepiNP$scHCL_label))
Idents(P02.TtsMepiNP) <- "scHCL_label"
# lable 통합 정리: 비슷한 cell type 하나로 모아서 highlight할 cell type 정리
Mucous.epithelial <- WhichCells(P02.TtsMepiNP, idents = c("Mucous.epithelial.cell_REG1A.high.Adult.Gall.Bladder1.","Gastric.mucosa.cell.Adult.Stomach1.","Gastric.mucosa.cell.Adult.Stomach3.","Mucous.epithelial.cell_TFF1.high.Adult.Gall.Bladder1."))
Pit <- WhichCells(P02.TtsMepiNP, idents = c("Pit.cell_WFDC21P.high.Adult.Stomach1.","Pit.cell_TFF2.high.Adult.Stomach1.","Pit.cell_TFF2.high.Adult.Stomach3.","Pit.cell_FOXQ1.high.Adult.Stomach1."))
Ductal <- WhichCells(P02.TtsMepiNP, idents = c("Ductal.cell.Adult.Pancreas_Baron.","Duct.cell.Adult.Pancreas_Muraro."))
Immune <- WhichCells(P02.TtsMepiNP, idents = c("Blood.NK.CD16..Placenta_VentoTormo.","T.cells1.Placenta_VentoTormo.","T.cells2.Placenta_VentoTormo.","Neutrophil_S100P.high.Adult.Omentum3."))
OM <- WhichCells(P02.TtsMepiNP, idents = c("Mesothelial.cell.Adult.Omentum2.","Granulocyte.Adult.Omentum3."))
#그림 그리기
DimPlot(P02.TtsMepiNP, label=F, group.by="scHCL_label", cells.highlight= list(Mucous.epithelial,Pit,Ductal,OM,Immune), cols.highlight = rainbow(5,s=0.7), cols= "grey")
ggsave("P02.TtsMepiNP.scHCL.png")
This post is licensed under CC BY 4.0 by the author.