Single-cell RNA-sequencing Analysis
기초적인 10X scRNAseq data 분석 방법
single cell data를 생산해서 분석하는 가장 기본적인 방법에 대해서 정리해보려 한다.
현재 가장 많은 연구자들이 생산하는 방식중 하나인 10X genomics사의 3’ seq / dual index solution 을 사용해서 prep 과정 거치고 illumina 사용해서 데이터 생산했다는 가정하에 진행해보려 한다.
분석 과정은 다음과 같이 진행된다.
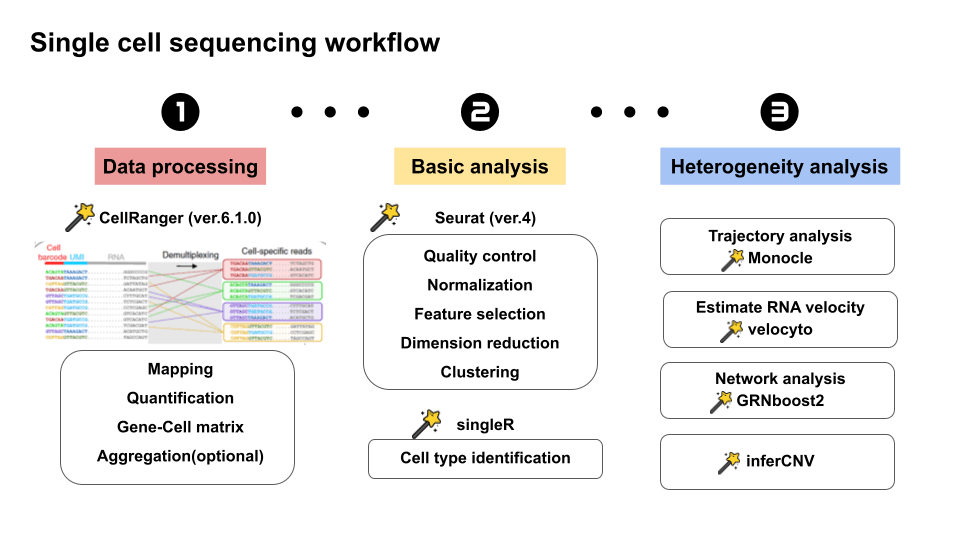
- CellRanger 단계: Read mapping & quantification
- Preprocessing 단계: Gene x cell count matrix를 R로 읽어 들여 분석.
Quality control 과정으로 low qual의 cell과 gene 제거 normalization -> feature selection -> dimension reduction -> clustering - 실험 목적에 맞는 다양한 분석 진행.
이 글에서는 본격적인 분석에 앞서 분석 준비하는 과정인 2번까지.
스탠다드 아닌 스탠다드 분석 방법을 정리해보고자 한다.
01. CellRanger
💎 Installation & Preparation
Cell Ranger download
여기에서 다운받을 수 있다. license agreement에 동의하고 약간의 개인정보를 입력해야 가능함.
CellRanger는 버전 업데이트가 진짜 빠른 편이다. 데이터 생산 할때마다 한번씩 확인해주는걸 추천한다.
2023 July 기준 최신 버전 7.1.0.
한 프로젝트의 데이터를 분석할때는 모든 샘플의 cellranger버전을 하나로 통일시키는것이 좋다.
설치 방법은 압축 풀고 경로 지정해서 바로 사용하면 된다.
1
wget -O cellranger-7.1.0.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-7.1.0.tar.gz?Expires=1689752445&Policy=eyJTdGF0ZW1lbnQiOlt7IlJlc291cmNlIjoiaHR0cHM6Ly9jZi4xMHhnZW5vbWljcy5jb20vcmVsZWFzZXMvY2VsbC1leHAvY2VsbHJhbmdlci03LjEuMC50YXIuZ3oiLCJDb25kaXRpb24iOnsiRGF0ZUxlc3NUaGFuIjp7IkFXUzpFcG9jaFRpbWUiOjE2ODk3NTI0NDV9fX1dfQ__&Signature=koD0oq0jj5bDsJNfJyHbz4e5LmmmvXikf80hz6h2j~GWwXzw7ccCr7KDAQ0UKonZ99cIQmB-dq1146xGGETEwExn8iGEKyggqpvBXhAILbXIF~QhomUTreQX9fTNQxSSjKg47NR8JTSztEjyex6rH7ir0Fsxpq2cqQXuXfmrM23PRZM2vLZTzo8i~AovapzwI54GwzdOAP9KH0RBt6AbonpDlwYHKyl7yz-fiypkerRTxC8LtDCCyKB~2a5nSWSYiGaCgW7nZ7oV1CKpDj64sDZLdyOaWRt0J1q0fjnrdbckngtlFDQCbVragnfBioUH46CAAks7Js2I2mbeUt20-A__&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA"
md5sum: 0938af789631800b20176527063c029a
Cell Ranger reference download
Cell Ranger를 돌리려면 reference가 필요한데, 직접 만들 수도 있고(custom reference), 10x genomics에서 미리 만들어 놓은 파일을 다운받아서 사용할수도 있다.
미리 제작해둔 reference 파일은 Cell Ranger 다운 받는 페이지에서 함께 받을 수 있고, reference는 Cell Ranger버전에 맞추지 않아도 사용할 수 있어 한 번 다운받아놓으면 새 버전의 Cell Ranger를 깔아도 같은 reference로 사용가능하다.
1
2
3
4
5
6
7
#Human reference (GRCh38)
wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2020-A.tar.gz
#md5sum: dfd654de39bff23917471e7fcc7a00cd
#Mouse reference (mm10)
wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-mm10-2020-A.tar.gz
#md5sum: 886eeddde8731ffb58552d0bb81f533d
human & mouse 함께 있는 버전도 있고, 각각의 build steps 링크를 눌러 들어가면 어떤 버전의 파일을 사용해서 만들었는지 과정을 확인할 수 있다.
💡 오른쪽 아래 빨간 네모가 있을텐데 누르면 이전 버전의 프로그램들도 다시 다운 받을 수 있다!
💎 Cell Ranger Count
가장 기본적인 CellRanger pipeline은 아래와 같다.
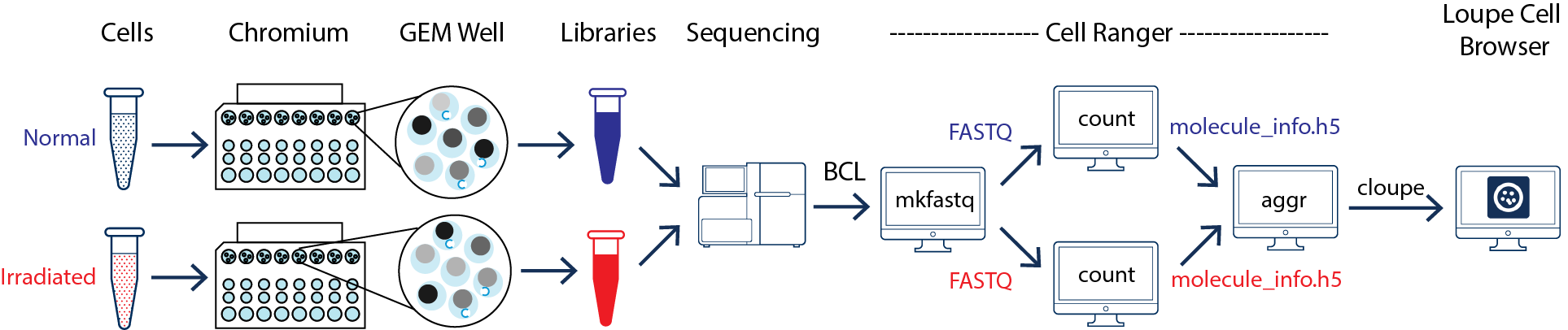 cellRanger Workflow Overview
cellRanger Workflow Overview
sequencing이 끝난 raw data는 Illumina BCL format으로 생산되는데, 분석을 시작하기 위해서는 FASTQ format으로 바꿔주는 과정이 필요하다. mkfastq 명령어를 사용하고, 과정 중에 필요하다면 demultiplex 과정을 거치기도 한다.
🍀 Demultiplexing
“Demultiplexing”이란, pooling과 반대되는 뜻으로 사용된다.
sampling pooling과정에서는 여러 sample에서 온 cell들에 cell barcode를 달아 origin을 표시하고 모든 cell을 한데 모아 한번에 sequencing했다면, Demultiplexing과정에서는 read들을 origin에 따라 분류해낸다.
Demultiplexing refers to the step in processing where you’d use the barcode information in order to know which sequences came from which samples after they had all be sequenced together.
대부분 데이터 생산은 회사에 의뢰하기 때문에 이 과정까지는 처리된 상태로 전달받는데, 만약 직접 하고 싶다면 견적을 받을때 요청하면 BCL상태로 데이터를 받아볼 수도 있다.
CellRanger count는
raw sequence data(.fastq)를 input으로 받아 reference genome에 mapping하고 quantificate해서 Gene x Cell의 gene expression matrix를 만들어준다.
이 글은 기본적인 RNA seq 데이터를 기준으로 하기 때문에 만약 Targeted Gene Expresssion 이나 Feature Barcode 데이터를 함께 생산했다면 링크를 따라 그에 맞는 pipeline을 사용하기 바란다.
기본적으로 Cell Ranger는 사용 가능한 모든 core와 memory를 끌어다 쓰기 때문에 제한할 목적이 아니라면 늘리기 위해서는 따로 옵션을 넣지 않아도 된다.
만약 받은 fastq 파일의 위치와 리스트가 다음과 같다고 하면..
1
2
3
4
5
6
$ cd /01_Data/outs/
$ ls
ProjectX_SampleName_S2_L001_I1_001.fastq
ProjectX_SampleName_S2_L001_I2_001.fastq
ProjectX_SampleName_S2_L001_R1_001.fastq
ProjectX_SampleName_S2_L001_R2_001.fastq
file name에서 “두번째 샘플(S2)에서 얻은 library를 첫번째 lane(L001)에 로딩해서 얻은 데이터라는 것과 dual index solution을 사용했음’‘을 알 수 있다.
이걸 input으로 cellranger count를 돌리려면
1
2
3
4
5
6
7
8
9
10
11
12
cd /02_count
id=SampleID01 # whatever you want
reference_dir=refdata-gex-GRCh38-2020-A # reference genome location
input_dir=01_Data/outs/ # where fastq storaged
fastq=ProjectX_SampleName
cellranger count --id=$id \
--transcriptome=$reference_dir \
--fastqs=$input_dir \
--sample=$fastq \
--nosecondary
input_dir에 여러 샘플의 데이터가 함께 있다면 --sample옵션을 반드시 넣어줘야한다. 아니라면 --fastqs만 잘 넣어주면 알아서 인식해서 돌아간다.
어차피 후속 분석을 직접 진행할 예정이기 때문에 --nosecondary 를 붙여주는것이 좋다. 좀 더 빨리 끝나고 output 파일 크기도 줄어든다.
- AMD 서버 32cores 256GB memory 기준 3,500 cells -> 1hr (tmp files 86GB) 22,000 cells -> 2hr (tmp files 102GB)
다 돌리고 나면 /02_count 아래에 지정한 id와 동일한 이름으로 폴더가 생기고 그 아래에 결과가 저장된다.
🍀 Output files
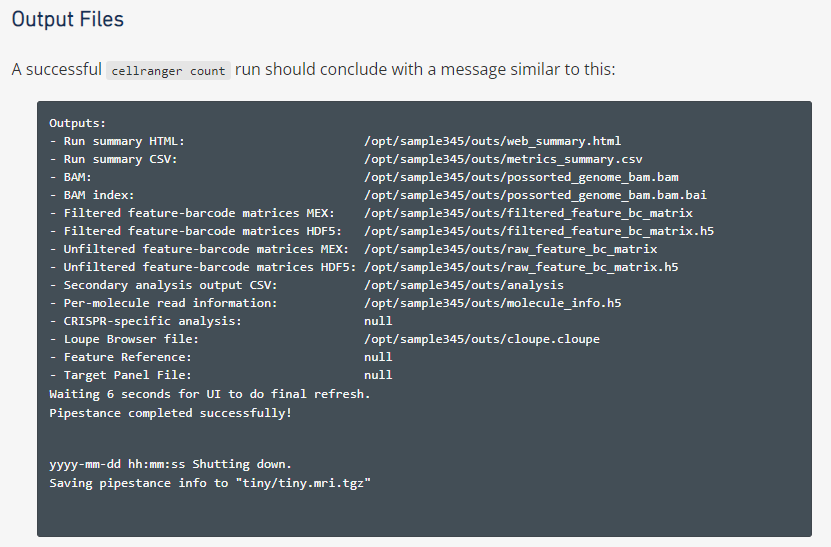

분석에 사용할 데이터들은 /outs 폴더 안에 들어있다.
web_summary.html
sequencing depth와 몇개 정도의 cell이 capture됐는지,count과정에 사용한 reference 버전 과 read quality, 발현 하는 평균 유전자의 갯수 등등을 정리해서 한눈에 확인 할 수 있다./filtered_feature_bc_matrix&raw_feature_bc_matrix
둘 폴더 모두 열어보면,
barcodes.tsv.gz,features.tsv.gz,matrix.mtx.gz가 들어 있다. raw 에는 empty droplet까지 포함한 모든 droplet에 대한 sequencing results가 들어 있고, filtered에는 cellranger에서 기본적으로 내장되어있는 기능으로 droplet을 걸러내고 어느정도 live cell이 capture 됐을 결과만 포함시킨 결과가 들어 있다. 어떤 파일을 쓸지는 본인의 선택. 물론 raw를 선택했다면 empty droplet을 걸러내는 quality control(QC) 과정을 더 빡세게 거쳐야겠지..molecule_info.h5aggr스텝을 거칠거라면 aggr.csv를 작성할때 넣는 파일
💎 Cell Ranger Aggr (optional)
여러 샘플을 한번에 분석한다면,.. aggr을 통해 하나로 합쳐주는 과정을 거쳐야한다.
돌리기 전에 aggr.csv라는 파일을 하나 만들어준다.
1
2
3
sample_id,molecule_h5
SampleID01,/02_count/SampleID01/outs/molecule_info.h5
SampleID02,/02_count/SampleID01/outs/molecule_info.h5
뿅!
1
cellranger aggr --id=Project_Name --csv=aggr.csv --nosecondary
하면 이전과 동일하게 Project_Name으로 폴더가 생기고, Project_name/outs/count/filtered_feature_bc_matrix 아래의 파일들을 후속 분석에 사용하면 된다.
02. Preprocessing with R (Seurat)
💎 R packages
이 이후의 과정은 R 을 이용해서 진행된다.
필요한 R package는 다음과 같고 설치 방법은 링크로 남겨두겠다.
1
2
3
4
5
6
7
library(dplyr)
library(Seurat)
library(patchwork)
library(tidyverse)
library(RColorBrewer)
library(ggplot2)
library(reticulate)
💎 Data loading
1
2
data <- Read10X(data.dir = "[CellRanger_results_dir/outs/filtered_feature_bc_matrix]")
SO <- CreateSeuratObject(counts = data, project = "[Project_name]",min.cells=10 )
만들어진 Seurat Object(SO)를 확인해보면..
1
2
3
4
> SO
An object of class Seurat
22063 features across 9065 samples within 1 assay
Active assay: RNA (22063 features, 0 variable features)
해석: 22063개의 유전자(feature/gene)에 대한, 9065개의 cell(sample)의 expression 정보가 있다.
Seurat object에는 앞으로 여러개의 assay가 등장 & 저장 될텐데,
가장 기본이 되는 assay가 RNA assay이다. 그 외, SCT, Integrated 등이 있다.
이 과정에서 여러 샘플을 한번에 분석하는 거라면,
다음 과정으로 넘어가기 전에 Labeling 해주면 좋다.
👉 cell ID에 라벨 달기
💎 Quality Control (QC)
가장 기본적인 QC단계로는 두 가지가 있다.
- Cell QC: 상태 안좋은 Cell 제거
- Gene QC: 발현 정도가 전반적으로 낮은 유전자 제거
Cell QC
상태가 안좋은 cell이라고 하면, 죽어가는 Cell을 의미한다.
일반적으로 붙어자라던 cell들은 single-cell sequencing 하기 위해 dissociation과정을 거치게 되면 apoptosis로 들어가게 되면서 사멸되는 경우가 대부분이기 때문에 이런 cell들은 앞으로의 분석에서 제외하는 것이 좋다.
Apoptosis 과정에 들어가게 되면 cell내에서 Mitochondrial(MT) gene 의 발현이 많아진다는 점을 이용. cell 마다 발현되는 유전자들 종류중에 MT gene의 %를 계산해서 상태가 안좋은(MT gene % 가 높은) cell들을 제거한다.
일반적으로 15~20% 언저리를 cutoff로 잡고 filtering 하는데, 확실하게 정해진 standard는 없다. scatter plot과 violin plot을 보며 경향을 보고 결정하는 것이 필요하다.
- 보통 유전자 200개미만으로 발현하는 cell들은 cell의 조각 또는 별 의미 없는 cell로 생각해서 제거하는데 주의!! 데이터의 종류에 따라 발현이 적은 특징이 있는 cell들이 포함돼 있을 가능성이 있다면 살려줘라. 하지만 보통은 제거함. 왜냐하면 나중에 feature selection과정에서 cell type들은 구분할 수 있는 유전자들을 기본옵션으로 3000개 뽑는데, 원래도 발현되는 유전자가 200개 밖에 없으면 noise처럼 작용될 수 있어서.
Gene QC
single cell expression data는 Gene X Cell의 matrix로 이루어져 있다.
유전자의 갯수는 (사람 기준) 약 2만여개로 정해져 있고, cell의 갯수로 데이터 사이즈가 결정되는데..
대부분의 데이터는 single-cell data특성상 굉장히 sparse하다.1
많게는 5000여개의 유전자 제외하고 나머지 유전자들의 발현은 0일 가능성이 굉장히 높음.
matrix를 의미도 없는 0으로 채워서 후속 분석까지 가져가는것은 자원 낭비 & 계산시간만 늘리는 악영향을 주기 때문에 대부분의 Cell에서 발현이 없는 유전자는 필터링해서 데이터 사이즈를 줄이는 것이 좋다.
이 필터링 과정은 CreateSeuratObject() 과정에서 min.cells 옵션으로 조정가능하다. 10개로 설정했다는 것은 cell 10개이상에서 발현되는 유전자만 갖고 오겠다는 뜻. 전체 cell중에 9개의 cell에서만 발현되는 유전자는 expression matrix에서 제외된다.
Data quality Checking code
1
2
3
4
5
6
7
### MT gene % 계산
SO[["percent.mt"]] <- PercentageFeatureSet(SO, pattern = "^MT-")
### Visualize QC metrics as a violin plot
png("00_filtering_QC_vln.png",width=1000, height=400)
VlnPlot(SO, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3, pt.size = 0)
dev.off()
없애기 전에 데이터의 전반적인 퀄리티를 확인해보자.
nFeature_RNA: cell별로 발현하는 유전자의 종류-
nCount_RNA: cell별로 총 UMI count값 이 두가지 값은 처음에 data loading단계에서CreateSeuratObject()하면 자동으로 meta data에 생성된다. percent.mt:PercentageFeatureSetfunction을 사용하면 MT gene %를 계산해준다. 이 값을 meta data에 “percent.mt”라는 이름으로 저장한 것.
사람은 “^MT-“(MT로 시작하는 유전자) / Mounse 의 경우는 “^mt-“ 를 사용한다.
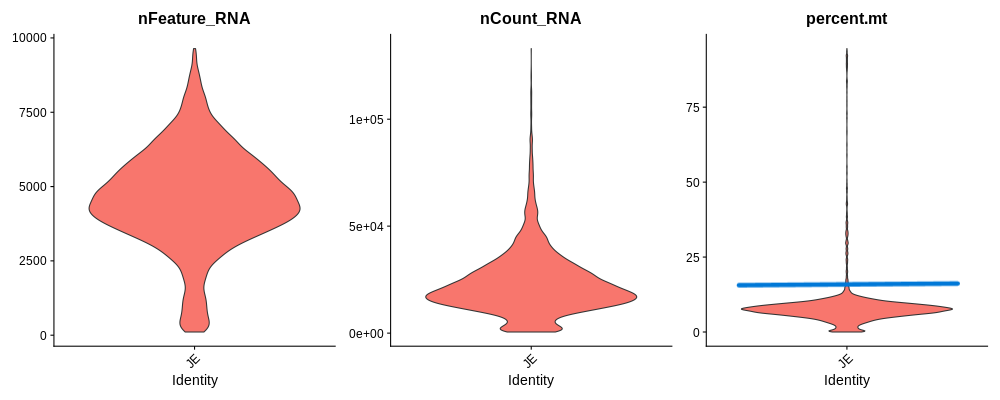 percent.mt의 cutoff를 어떻게 잡아야하는가?
percent.mt의 cutoff를 어떻게 잡아야하는가?
15~25%사이 중 outlier로 여겨질 정도의 선을 잡는다. like the blue line. 애매하쥬? 하지만 정해진 규칙은 따로 없으니 정말 죽어가는 애들을 배제할 정도로 잡는다고 생각하면 편함. 너무 naive하게 잡아서 (30%) 상태가 안좋은 cell들까지 분석에 포함시키면 나중에 걔들끼리 따로 clsuter를 형성할 수도 있음. 그러면 그런 애들을 제거하려고 일을 두번해야할수도 있다.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
# filter되는 cell과 남길 cell들에 label을 달면 QC결과 정리에 도움이 됨
SO@meta.data$MTQC <- "PASS"
SO@meta.data[colnames(x= subset(x = SO, subset = percent.mt >= 15)),'MTQC'] <- "Filtered"
#> table(LLC$MTQC)
SO@meta.data$FCQC <- "PASS"
SO@meta.data[colnames(x= subset(x = SO, subset = nFeature_RNA < 200 )),'FCQC'] <- "Filtered"
#> table(LLC$FCQC)
plot1 <- FeatureScatter(SO, feature1 = "nCount_RNA", feature2 = "percent.mt",pt.size = 0.01, group.by="MTQC")
plot2 <- FeatureScatter(SO, feature1 = "nCount_RNA", feature2 = "nFeature_RNA",pt.size = 0.01, group.by="FCQC")
png("00_filtering_QC_scat.png",width=800, height=400)
wrap_plots(plot1,plot2)
dev.off()
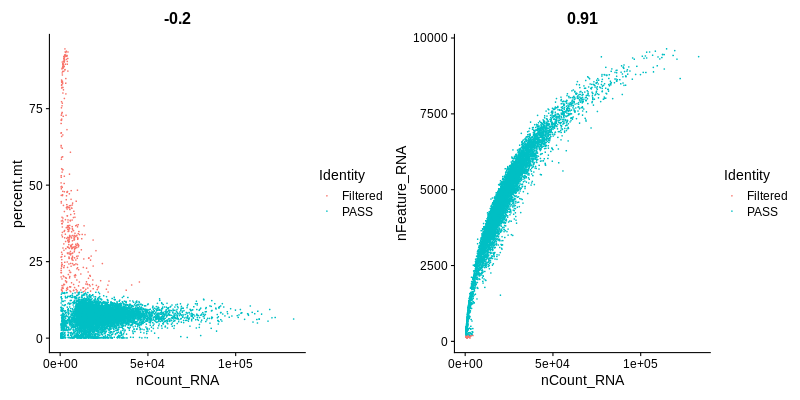 Scatter plot으로 그려봤을때, percent.mt 기준으로 제거되는 cell들이 이런 모양이면 좋다. MT gene %도 높고 전체적인 발현은 낮은 cell들만 제거되면 됨.(왼쪽 위로 길게 올라가는 cell들 제거. 아랫쪽에 뭉쳐 있는 부분은 살리고) // Feature count를 기준으로 너무 feature가 많은(8~9천 gene 이상) cell들이 outlier처럼 있다면 doublet일 가능성이 있다. 제거해야한다고 생각되면 제거 하는 것도 좋다 OR 일단은 살려두고 doublet detector를 사용하는 것도 하나의 방법.
Scatter plot으로 그려봤을때, percent.mt 기준으로 제거되는 cell들이 이런 모양이면 좋다. MT gene %도 높고 전체적인 발현은 낮은 cell들만 제거되면 됨.(왼쪽 위로 길게 올라가는 cell들 제거. 아랫쪽에 뭉쳐 있는 부분은 살리고) // Feature count를 기준으로 너무 feature가 많은(8~9천 gene 이상) cell들이 outlier처럼 있다면 doublet일 가능성이 있다. 제거해야한다고 생각되면 제거 하는 것도 좋다 OR 일단은 살려두고 doublet detector를 사용하는 것도 하나의 방법.
Data filtering code
1
2
SO <- subset(SO, subset = percent.mt < 15)
SO <- subset(SO, subset = nFeature_RNA >= 200)
💎 Normalization, Feature selection, Dimension reduction
Normalizaiton
Good quality cell만 남겼다면, Normalization해준다.
single-cell data의 zero-inflated data를 다루기엔 scTransform normalization이 제격이지만, 나중에 Differentailly Expressed Genes(DEG)도 구해야하고 heatmap그리려면 log-normalized (RNA assay)도 필요하기 때문에 둘 다 해줄거다.
1
2
3
4
5
6
7
# Log norm -> 결과는 RNA assay에 저장된다.
SO <- NormalizeData(SO)
all.genes <- rownames(SO)
SO <- ScaleData(SO, features = all.genes)
# SCTransform -> 결과는 SCT assay에 저장된다.
SO <- SCTransform(SO, vst.flavor = "v2")
SCTransform function 안에는 RNA assay의 NormalizeData(), ScaleData(), FindVariableFeatures()과정이 모두 포함되어 있다.
Dimension reduction & Clustering
지금 data는 multi-dimension 상태(2만여개의 Gene x 수만개 cell). Visualization시키기 위해서는 dimension reduction이 필요하다.
1
2
3
4
SO <- RunPCA(SO, verbose = FALSE)
ElbowPlot(SO, ndims=50)
ggsave("02.SO.elbow.png")
RNA assay였다면 jackstraw plot을 함께 그려서 몇개의 PC를 사용할지 쉽게 정할 수 있겠지만, SCT assay에서는 Jackstraw를 돌릴 수 없다.
하지만 SCTransform은 어느정도 range의 PC를 커버 할 수 있어서.. 정확한 elbow point를 찍지 않고 좀 더 잡아도 괜찮다.. 고 seurat vignette에 적혀 있음.2
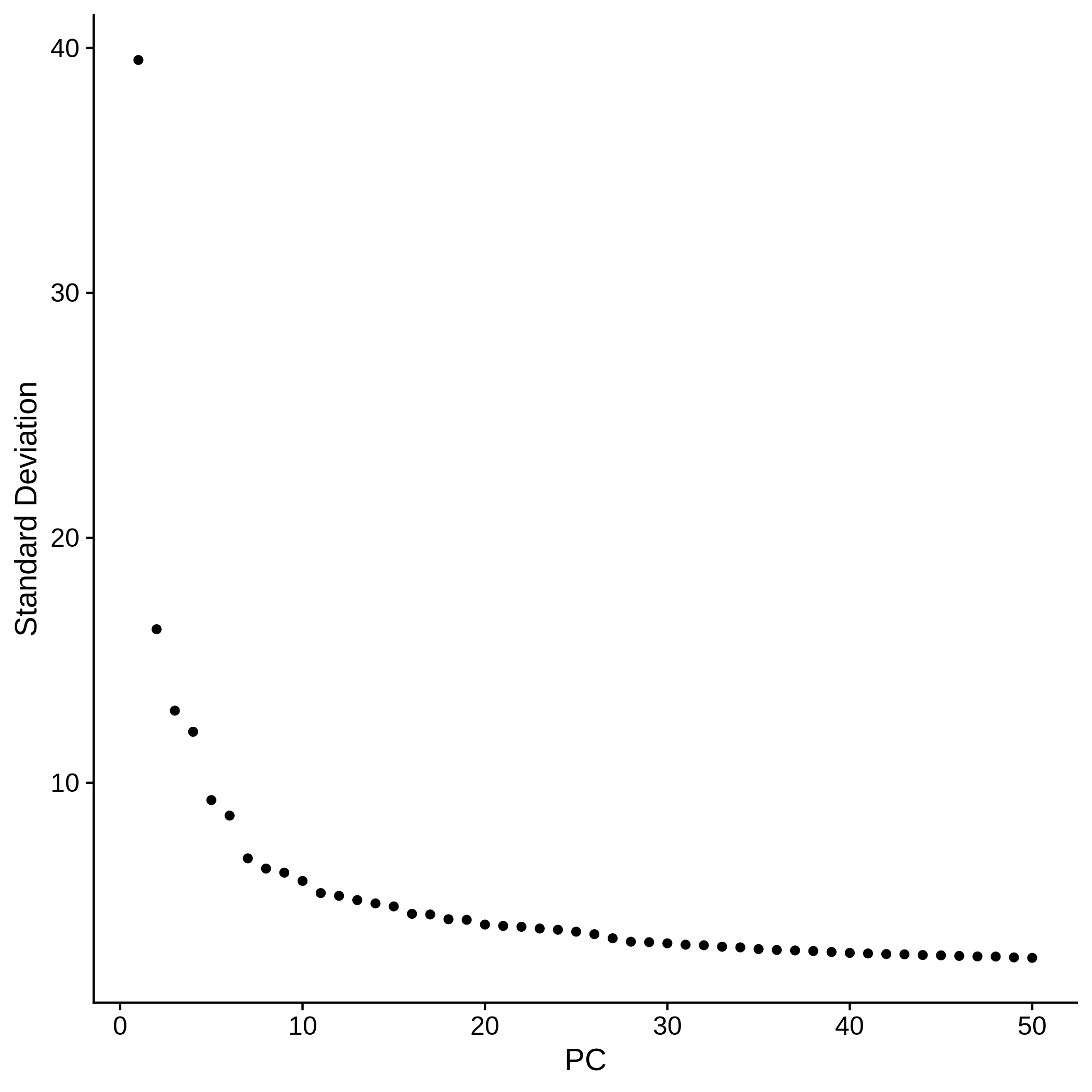 정확한 elbow point는 7~8이겠지만 난 PC로 10까지 잡겠다. benefit of SCT
정확한 elbow point는 7~8이겠지만 난 PC로 10까지 잡겠다. benefit of SCT
1
2
3
4
5
6
7
8
9
PC=10
#SO <- RunTSNE(SO, dims = 1:PC) # tSNE로 dimension reduction 요즘은 잘 안쓴다.
SO <- RunUMAP(SO, dims = 1:PC, reduction = "pca", umap.method="umap-learn")
# umap-learn 메소드 사용하려면 따로 설치해줘야 함 (하나 골라서)
## reticulate::py_install(packages ='umap-learn')
## pip install umap-learn
## conda install -c conda-forge umap-learn
Clustring & Visualization
Clustering하고 그림으로 그려보자.
1
2
3
4
5
SO <- FindNeighbors(SO, dims = 1:PC,reduction = "pca", verbose = FALSE)
SO <- FindClusters(SO, resolution = 0.1, verbose = FALSE)
DimPlot(JE, reduction='umap', label=TRUE) + NoLegend()
ggsave("02.SO.umap_0.1_PC10.png")
FindCluster()의 resolution을 조절하면 cluster갯수를 조절 할 수 있다.
숫자가 작아질수록 cluster의 갯수도 작아진다. 커지면 커질수록 자잘하게 나눠줌.
UMAP모양 자체를 바꾸고 싶다면 PC를 조절해야한다.
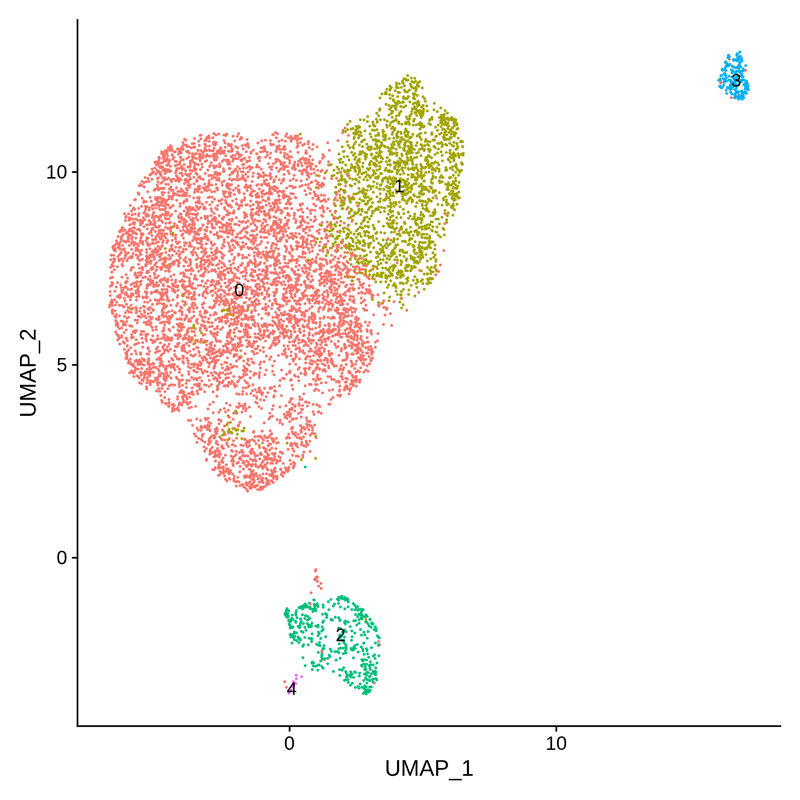
자 이제 Cluster별로 cell type을 찾아 나서면 가장 기본적인 분석이 끝난다!
Cell type annotation은 또 멀고 긴 길이니 다음 글에서..
이글에서 사용한 R sessionInfo
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
> sessionInfo()
R version 4.1.1 (2021-08-10)
Platform: x86_64-conda-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /home/subin/.conda/envs/Project13/lib/libopenblasp-r0.3.18.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] reticulate_1.22 RColorBrewer_1.1-2 forcats_0.5.1 stringr_1.4.0
[5] purrr_0.3.4 readr_2.1.0 tidyr_1.1.4 tibble_3.1.6
[9] ggplot2_3.3.5 tidyverse_1.3.1 patchwork_1.1.1 SeuratObject_4.0.3
[13] Seurat_4.0.5 dplyr_1.0.7
loaded via a namespace (and not attached):
[1] Rtsne_0.15 colorspace_2.0-2 deldir_1.0-6
[4] ellipsis_0.3.2 ggridges_0.5.3 fs_1.5.0
[7] rstudioapi_0.13 spatstat.data_2.1-0 farver_2.1.0
[10] leiden_0.3.9 listenv_0.8.0 ggrepel_0.9.1
[13] fansi_0.4.2 lubridate_1.8.0 xml2_1.3.2
[16] codetools_0.2-18 splines_4.1.1 polyclip_1.10-0
[19] jsonlite_1.7.2 broom_0.7.10 ica_1.0-2
[22] cluster_2.1.2 dbplyr_2.1.1 png_0.1-7
[25] uwot_0.1.10 shiny_1.7.1 sctransform_0.3.2
[28] spatstat.sparse_2.0-0 compiler_4.1.1 httr_1.4.2
[31] backports_1.3.0 assertthat_0.2.1 Matrix_1.3-4
[34] fastmap_1.1.0 lazyeval_0.2.2 cli_3.1.0
[37] later_1.2.0 htmltools_0.5.2 tools_4.1.1
[40] igraph_1.2.8 gtable_0.3.0 glue_1.5.0
[43] RANN_2.6.1 reshape2_1.4.4 Rcpp_1.0.7
[46] scattermore_0.7 cellranger_1.1.0 vctrs_0.3.8
[49] nlme_3.1-153 lmtest_0.9-39 globals_0.14.0
[52] rvest_1.0.2 mime_0.12 miniUI_0.1.1.1
[55] lifecycle_1.0.1 irlba_2.3.3 goftest_1.2-3
[58] future_1.23.0 MASS_7.3-54 zoo_1.8-9
[61] scales_1.1.1 spatstat.core_2.3-1 hms_1.1.1
[64] promises_1.2.0.1 spatstat.utils_2.2-0 parallel_4.1.1
[67] pbapply_1.5-0 gridExtra_2.3 rpart_4.1-15
[70] stringi_1.7.5 rlang_0.4.12 pkgconfig_2.0.3
[73] matrixStats_0.61.0 lattice_0.20-45 ROCR_1.0-11
[76] tensor_1.5 labeling_0.4.2 htmlwidgets_1.5.4
[79] cowplot_1.1.1 tidyselect_1.1.1 parallelly_1.28.1
[82] RcppAnnoy_0.0.19 plyr_1.8.6 magrittr_2.0.1
[85] R6_2.5.1 generics_0.1.1 DBI_1.1.1
[88] pillar_1.6.4 haven_2.4.3 withr_2.4.2
[91] mgcv_1.8-38 fitdistrplus_1.1-6 survival_3.2-13
[94] abind_1.4-5 future.apply_1.8.1 modelr_0.1.8
[97] crayon_1.4.2 KernSmooth_2.23-20 utf8_1.2.2
[100] spatstat.geom_2.3-0 plotly_4.10.0 tzdb_0.2.0
[103] grid_4.1.1 readxl_1.3.1 data.table_1.14.2
[106] reprex_2.0.1 digest_0.6.28 xtable_1.8-4
[109] httpuv_1.6.3 munsell_0.5.0 viridisLite_0.4.0
참고할만한 페이지
- Bioconductor: Single-cell analysis toolkit for expression in R
- BioCellgen: processing scRNA-seq data with Seurat
- Analyzing single-cell RNA-seq data containing UMI counts
-
single-cell data가 sparse해지는 이유로는 ↩