[10x/Seurat] 여러 종 섞어만든 scRNA-seq 데이터에서 종별 cell 구분하는 법
cancer study에서 많이 사용하는 PDX로 scRNA-seq data를 만들면 그 안에는 human cell(암덩이)과 mouse cell(그안에 들어간 immune cell etc.)이 함께 들어있기 마련이다.
제대로 분석하기 위해서는 이 두 종에서 온 cell들을 구분지어서 따로 분석할 필요가 있는데, 이렇게 하기 위해서는
먼저 mapping단계에서 (CellRanger) 두 종의 reference genome을 모두 사용해서 분석해야한다.
CellRanger reference 중에 human(GRCh38) 과 mouse(mm10) reference를 하나로 합쳐놓은 버전이 있으니 PDX 데이터는 이 reference를 사용해서 CellRanger count 진행하면 된다.
- 👉 CellRanger reference: human(GRCh38) + mouse(mm10) download
wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-and-mm10-2020-A.tar.gz
다른 종 조합 reference 만드는법
두번째로 만들어진 gene/cell count matrix에서 cell을 어떤 종에서 유래했는지 알아내서 나눠야하는데..
UMI count 비율을 사용해 종별로 cell 나누기 in R
UMI count를 사용해서 각 cell별로 어떤 종의 reference에 더 많이 mapping됐는지 확인하고 그 정도에 따라서 나눠보자.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
library(Seurat)
options(Seurat.object.assay.version = "v5")
library(ggplot2)
library(patchwork)
library(tidyverse)
# 10X CellRanger count 결과 load
PDX.data <- Read10X("CellRanger/outs/filtered_feature_bc_matrix")
###01.human vs mouse
# gene name에 GRCh38이라는 패턴의 여부에 따라 human과 mouse gene을 나누고 labeling
gene_species <- ifelse(str_detect(rownames(PDX.data), "^GRCh38"), "human", "mouse")
mouse_inds <- gene_species == "mouse"
human_inds <- gene_species == "human"
#table(gene_species)
#gene_species
#human mouse
#33538 31053
# mark cells as mouse or human
cell_species <- tibble(n_mouse_umi = Matrix::colSums(PDX.data[mouse_inds,]),
n_human_umi = Matrix::colSums(PDX.data[human_inds,]),
tot_umi = Matrix::colSums(PDX.data),
prop_mouse = n_mouse_umi / tot_umi,
prop_human = n_human_umi / tot_umi)
cell_species<- cell_species %>%
mutate(species = case_when(
prop_mouse > 0.9 ~ "mouse", # 나는 90%이 각 종에서 온 UMI면 그쪽 종 cell로 정의했다.
prop_human > 0.9 ~ "human", # banyard plot을 그려보고 본인 case에 맞게 조정하자.
TRUE ~ "mixed"
))
# labeling
h_cell <- colnames(PDX.data)[cell_species$species=="human"]
m_cell <- colnames(PDX.data)[cell_species$species=="mouse"]
#length(h_cell[grepl("(^(\\w*)-1$)",h_cell)])
# 종 별로 데이터 나누고 정리
PDX.human.data <- PDX.data[rownames(PDX.data)[human_inds], cell_species$species=="human"]
rownames(PDX.human.data) <- gsub(rownames(PDX.human.data),pattern="GRCh38_", replacement="")
dim(PDX.human.data)
#[1] 33538 22342
saveRDS(PDX.human.data,"01.PDX.human.data.rds")
PDX.mouse.data <- PDX.data[rownames(PDX.data)[mouse_inds], cell_species$species=="mouse"]
rownames(PDX.mouse.data) <- gsub(rownames(PDX.mouse.data),pattern="mm10___", replacement="")
dim(PDX.mouse.data)
#[1] 31053 19437
saveRDS(PDX.mouse.data,"01.PDX.mouse.data.rds")
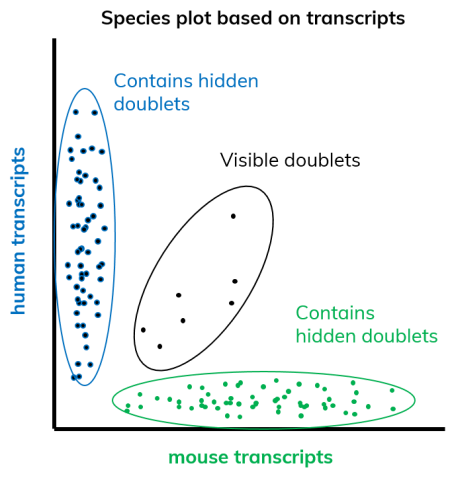
Barnyard plot
위의 코드에서 봤듯이 어느정도의 UMI가 어떤 종에서 유래했는지. 그 정도에 따라서 cell들이 갈리기 때문에 몇%로 잡을건지가 중요하다.
보통은 90~95%가 많이 보이고 (물론 높으면 높을수록 좋겠지!)
기준을 그냥 잡기 어렵다면 barnyard plot을 그려보자.
보통은 technical doublet rate을 알아보기 위해서 일부러 두 종의 cell을 섞고 이 그림을 그려보는데,..
우리는 그려서 [종별 UMI count]/[total UMI count] 비율의 기준을 정할때 사용할 거다.
1
2
3
4
ggplot(cell_species, aes(x=n_human_umi, y=n_dog_umi,color=species)) +
geom_point() +
scale_color_manual(values=c('#FF4C65','#06B2F8','#8946DE'))
ggsave("banyard95.png")
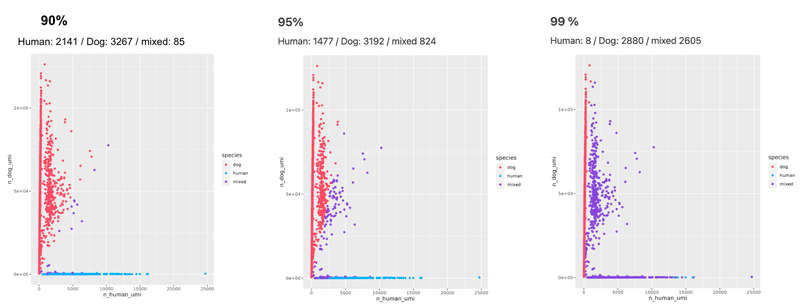 이런식으로 여러 %로 그려보고 결정하자
이런식으로 여러 %로 그려보고 결정하자
위 프로젝트에서는 human cell만 얻으려고 했던거라 난 95%를 선택했다.
끝