MEGENA 튜토리얼: 설치부터 기본 사용, visualization까지
MEGENA: Multiscale embedded gene co-expression network analysis
논문 정보
Song, Won-Min, and Bin Zhang. “Multiscale embedded gene co-expression network analysis.” PLoS computational biology 11.11 (2015): e1004574. pdf download
Installation
Prerequisite
-
- R version > 4.3
- dependency package
impute, bioconductor ver.3.17부터 가능한데 이게 R 4.3부터 가능
impute
1
BiocManager::install("impute")
intall MEGENA
1
2
3
4
library(devtools);
install_github("songw01/MEGENA");
# OR
install.packages("MEGENA") #이거
⚠️ 아직 계속해서 업데이트중
최신버전 사용하고 싶으면, github코드로 사용
How to run MEGENA
Prepare inputs
1
2
3
4
5
6
7
8
9
10
11
12
13
library(Seurat)
options(Seurat.object.assay.version = "v5")
GEX.seurOBJ <- readRDS("GEX.seurOBJ_annotated.rds")
# by celltypes
for(i in levels(GEX.seurOBJ$Annot.L2)){
celltype.obj <- subset(GEX.seurOBJ,subset=Annot.L2==i)
data_matrix <- celltype.obj[["RNA"]]$data
data_matrix <- data_matrix[rowSums(data_matrix)>0,]
#data_matrix <- LayerData(celltype.obj, assay="RNA", layer="data")
write.table(data_matrix, file=paste0('data_matrix_',i,'.tsv'), quote=FALSE, sep='\t')
}
MEGENA input으로 준비하는 matrix에 대해서,..
RNA vs. SCT assay? Counts vs Data layer?
어떤 데이터를 사용해야할지 고민이 많았는데, 결론부터 말하면 어떤 것을 쓰던 결과는 동일하게 나온다
assy 종류, layer 종류 모두 상관없음.
run MEGENA
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
cellType_list <- c("CD4 T","CD8 T","Proliferating T","NK","B","Plasma","CD14 Monocyte","CD16 Monocyte","Monocyte","cDC","pDC")
library(MEGENA)
anyNA <- function(x) any(is.na(x))
source("recap_source.R")
source("check_matrix.R")
wk <- "/sc/arion/projects/scMultiscale/chos14/Project01_sysFlu/04_MEGENA/" # working directory within MEGENA directory
args=commandArgs(trailingOnly=TRUE)
FDR.cutoff <- 0.5
q.pr <- 0.05 # filter of low responding probes based on quantiles
n.cores <- 16
# input parameters
doPar <- TRUE;
method = "spearman"
#FDR.cutoff = 0.05
module.pval = 0.05
hub.pval = 0.05
#################################################################
i = cellType_list[as.numeric(args[1])]
print(as.numeric(args[1]))
setwd(wk)
df <- paste0("woP03_data_matrix_",i,".tsv") # Expression file, rows: probes; colums: samples; 1st col: probe-names
print(df)
# variable required: df, wk, FDR.cutoff, n.cores
data.file <- df # presuming that we use the full file name
annot.col <- 1 ## number of annotation columns in the data matrix
annot <- 1 ## column with annotation to be used
wkdir <- paste0(wk,i,"_FDR",FDR.cutoff,"mpi")
###########
# create output folder
dir.create(wkdir)
# load data
data.Df <- read.delim(file = data.file,sep = "\t",header = T)
data.mat <- as.matrix(data.Df);
v <- cv.calculation(data.mat)
res <- filter_low_quality_genes(data.mat,min.read = 1E-320,min.cv = quantile(v,probs=q.pr),min.sprop = 0.05)
datExpr <- res$expr.matrix
message("\n\nPrint some statistics:")
range(v)
message("Quantile prob: ", q.pr)
quantile(v,probs=q.pr)
dim(data.mat)
dim(datExpr)
# set working folder
setwd(wkdir)
#####
# calculate correlation
message("Calculate correlation")
set.seed(1234)
# with 0
# rho.out = calculate.rho.signed(datExpr,n.perm = 10,FDR.cutoff = 0.2,estimator = "pearson",
# use.obs = "na.or.complete",direction = "absolute",
# rho.thresh = NULL,sort.el = TRUE)
# without 0
rho.out = calculate.rho.signed(datExpr,n.perm = 10,FDR.cutoff = 0.2,estimator = "pearson",
use.obs = "pairwise.complete.obs",direction = "absolute",
rho.thresh = NULL,sort.el = TRUE)
ijw = subset(rho.out$signif.ijw, rho > 0.2)
write.table(ijw,paste0("ijw_",i,".txt"),,sep = "\t",row.names = F,col.names = T,quote = F)
message("Calculate PFN")
if (doPar & getDoParWorkers() == 1){
cl <- parallel::makeCluster(n.cores)
registerDoParallel(cl)
# check how many workers are there
cat(paste("number of cores to use:",getDoParWorkers(),"\n",sep = ""))
}
# calculate PFN (no)
el <- calculate.PFN(ijw[,1:3],doPar = doPar,num.cores = n.cores)
message("Write files")
write.table(el,file = paste0("MEGENA_Network_",i,".txt"),sep = "\t",row.names = F,col.names = T,quote = F)
# do clustering
g <- graph.data.frame(el,directed = F)
MEGENA.output <- do.MEGENA(g,
mod.pval = module.pval,hub.pval = hub.pval,remove.unsig = TRUE,
min.size = 10,max.size = vcount(g)/2,
doPar = TRUE,num.cores = n.cores,n.perm = 100,
save.output = TRUE)
m <- MEGENA.ModuleSummary(MEGENA.output, max.size=50000)
output.geneSet.file(m$modules, "multiscale_significant.modules.txt")
write.table(MEGENA.output$node.summary, file="multiscale_nodeSummary.txt", row.names=F, quote=F, sep="\t")
save(MEGENA.output,file = paste0("MEGENA_output_",i,".RData"))
rm("MEGENA.output","g","el","ijw","m")
이부분은 optional, server에 parallel job으로 넣고 싶어서 작성함.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
#BSUB -J megena_sc[1-11]
#BSUB -n 24
#BSUB -L /bin/bash
#BSUB -a openmpi
#BSUB -q premium
#BSUB -P acc_scMultiscale
#BSUB -R rusage[mem=100000]
#BSUB -R himem
#BSUB -W 96:00
#BSUB -o %J.out.%I
source ~/.bash_profile
cd $LS_SUBCWD
echo $LS_SUBCWD
module load openmpi
#get the job index of an array job
jobID=$LSB_JOBID
#run script
export R_LIBS_USER=/sc/arion/projects/scMultiscale/chos14/source/R-4.3.1/library
Rscript /sc/arion/projects/scMultiscale/chos14/Project01_sysFlu/04_MEGENA/megen
a.R ${LSB_JOBINDEX}
date
Outputs
중간에 만든 wkdir 에 가보면
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
.
├── cluster_stats.png
├── * correlation_FDR_table.txt
├── Data_Correlation.txt
├── ijw_CD4\ T.txt
├── majority_vote.png
├── * MEGENA_Network_CD4\ T.txt
├── MEGENA_output_CD4\ T.RData
├── multiscale_clusters.RData
├── multiscale_hubAnalysis.RData
├── multiscale_nodeSummary.txt
├── * multiscale_significant.modules.txt
├── pfg_el.RData
├── pvalue_distributions.png
├── scalecluster_heatmap.png
└── scalewise_hub_tileplot.png
* 표시한 파알들 주목.
주의사항
-
filter_low_quality_genes()이후 ngenes: xxx 갯수 확인.- 적어도 몇천 genes는 되어야 괜찮은 사이즈의 module을 얻을 수 있음.
-
correlation_FDR_table.txt- FDR값이 계속 decreasing인지 확인. 중간에 다시 올라가진 않는지 확인하자.
-
MEGENA_Network.txt- weight == correlation coefficient, 두 유전자가 얼마만큼의 correlation을 갖고 co-expressed되는지 알 수 있다. highest value가 어느정도인지 확인하자.
-
multiscale_significant.modules.txt- 최종 결과로 나온 significant module. 적어도 100개 이상이어야..
Downstream analysis
DEG enrichment
Functions
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
readSigModules <- function(mfile) {
x <- scan(mfile, what="", sep="\n")
y<-strsplit(x,"\t")
names(y)<-sapply(y, `[[`, 1)
y<-lapply(y, `[`, -1) # y is now module list
return(y)
}
fisherTest <- function(poplisthits, minPopulationHits=5){
if ( (poplisthits[2]<minPopulationHits) | (poplisthits[4]==0) )
return (1)
#old way, maybe wrong
#a=rbind( c(poplisthits[2]-poplisthits[4], poplisthits[1]-poplisthits[2]-poplisthits[3]+poplisthits[4]),
# c(poplisthits[4], poplisthits[3]-poplisthits[4] ) )
#cat(as.character(poplisthits[1]), as.character(poplisthits[2]),as.character(poplisthits[3]),as.character(poplisthits[4]),"\n")
#signif(fisher.test(a)$p.value,3)
q= poplisthits[4] #list hit
m= poplisthits[2] #population hit
n= poplisthits[1]-poplisthits[2] #population non-hit
k= poplisthits[3] #list total
#myp=1-phyper(q-1, m, n, k)
myp=phyper(q-1, m, n, k, lower.tail=F)
signif(myp,3)}
rankOrder <- function(p) {
r <- apply(p, 2, rank, ties.method="min")
g <- apply(r, 2, function(x) (max(x)+1-x)/sum(x))
G <- apply(g, 1, prod)
rnk <- length(G) - rank(G, ties.method="max") + 1
return(rnk)}
coexEnrich <- function(gn, d, bg = NULL, fc.thresh = 0, pvalCut = NULL) {
if (is.null(bg)) { bg <- unique(unlist(gn)) }
if (is.null(pvalCut)) { pvalCut = 0.05 }
N <- length(unique(bg))
X <- length(intersect(unique(bg), d))
out <- NULL
for (m in 1:length(gn)) {
n <- length(unique(gn[[m]]))
x <- length(intersect(gn[[m]], d))
message("Processing ", names(gn)[m], ": ", n, ",", x)
fc <- (x / n) / (X / N)
genes <- paste(intersect(gn[[m]], d), collapse = " ")
hgd <- fisherTest(c(N, X, n, x))
if (!is.nan(fc)) {
if (fc >= fc.thresh) {
o <- c(names(gn)[m], x, n, X, N, fc, hgd, genes)
}
} else {
o <- c(m, NA, NA, NA, NA, NA, NA, NA)
}
out <- rbind(out, o)
}
p.val <- as.numeric(out[, 7]) # p.value
all.pws <- length(which(as.numeric(out[, 2]) != 0)) # x
corr.p.val <- p.adjust(p.val, method = "fdr")
out <- cbind(out[, -8], corr.p.val, out[, 8])
colnames(out) <- c("Module", "x", "n", "X", "N", "fold.change", "p.value", "corr.p.val", "Genes")
return(out)
}
Enrichment analysis code
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
## DEG 뽑아놓고
# MEGENA results
output.summary <- readRDS("../Orig/MEGENA_Mono_wSC_wo0_g50/MEGENA.summary.RDS")
# DEG results
DEG.table <- read.table("../../Mono_DEG/Mono.WilcoxDEG.final.txt")
y = readSigModules("../Orig/MEGENA_Mono_wSC_wo0_g50/multiscale_significant.modules.txt")
# saveRDS(rownames(Mono), "bg.rds") # background genes
bg <- readRDS("bg.rds")
# module name MXX로 바꾸고 싶어서
mdf= output.summary$module.table
mdf$module.id <- sub("c1_", "M", mdf$module.id)
mdf$module.parent <- sub("c1_", "M", mdf$module.parent)
rownames(mdf) <- sub("c1_", "M",rownames(mdf))
## DEG enrichment
tm.DEG <- NULL
for (d in DEGs) { # use DEGs list
o <- coexEnrich(y, d, bg)
tm.DEG <- cbind(tm.DEG, o)}
cp <- apply(tm.DEG[,grep("p.value", colnames(tm.DEG))], 2, as.numeric)
ix<-apply(cp, 2, function(x) all(is.na(x)))
rnk <- rankOrder(cp[,which(!ix)])
colnames(cp) <- names(DEGs)
module <- sub("c1_", "M", tm.DEG[,1])
deg_enrichment_df <- cbind(tm.DEG[,1], module, rnk, cp) # cannot include cp matrix as # of DEGs differ
#write.table(deg_enrichment_df, file="DEGenrichment.g50.pval.txt", row.names=F, quote=F, sep="\t")
#deg_enrichment_df <- as.data.frame(deg_enrichment_df, stringsAsFactors = FALSE)
Sunburst plot 으로 visualization
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
# 3번째 layer 이후로는 글씨 크기 줄이고 싶어서 수정
draw_sunburst_wt_fill <- function(module.df,# module table
### main inputs to specify hierarchy
parent.col = "module.parent",# module parent column
id.col = "id",# module id columnm
min.angle = 5,# minimum angle of section to label for module id
# fill aspects
feat.col,# feature column (character) to color sunburst
fill.type = "continuous",# continuous/discrete, is the variable numeric or factor?
log.transform = TRUE,# TRUE/FALSE, do log10 transform for p-values?
fill.scale = NULL,
theme.adjust = NULL)
{
require(ggraph)
#############
# make ggraph object
hier.df = module.df[,c(parent.col,id.col)]
htree = graph_from_data_frame(hier.df,vertices = union(hier.df[[1]],hier.df[[2]]))
## create initial sunburst
sunb = ggraph(htree, 'partition', circular = TRUE)
# filling aspect
if (!is.null(feat.col))
{
if (fill.type == "continuous")
{
# process
vec = module.df[[which(colnames(module.df) == feat.col)]]
fill.lab = feat.col
if (!is.numeric(vec)) stop(paste("variable:",feat.col," is not numeric.",sep = ""))
if (log.transform)
{
vec = -log10(vec);
fill.lab = paste("-log10(",feat.col,")",sep = "")
}
sunb$data$fill.value = vec[match(sunb$data$name,module.df[[which(colnames(module.df) == id.col)]])]
sunb = sunb + geom_node_arc_bar(aes(fill = fill.value),linewidth = 0.25) +
guides(fill = guide_colorbar(title = fill.lab)) + fill.scale
}
if (fill.type == "discrete")
{
vec = module.df[[which(colnames(module.df) == feat.col)]]
fill.lab = feat.col
if (!is.factor(vec)) stop(paste("variable:",feat.col," is not factor.",sep = ""))
sunb$data$fill.value = vec[match(sunb$data$name,module.df[[which(colnames(module.df) == id.col)]])]
sunb = sunb + geom_node_arc_bar(aes(fill = fill.value),linewidth = 0.25) +
guides(fill = guide_legend(title = fill.lab)) + fill.scale
}
}else{
sunb = sunb + geom_node_arc_bar()
}
# add module id
# add label rotation feature
sunb$data$rotation = 180 - (90 + (sunb$data$start + sunb$data$end )/2 * (180/pi) ) - 90
sunb$data$rotation[sunb$data$depth == 0] = 0
sunb$data$sb.angle = abs(sunb$data$end - sunb$data$start) * (180/pi)
sunb$data$rotation[which(sunb$data$sb.angle < 10)] = sunb$data$rotation[which(sunb$data$sb.angle < 10)] + 90
sunb$data$font.size <- ifelse(sunb$data$depth <= 2, 4, 2)
sunb = sunb + geom_text(data = subset(sunb$data, sb.angle >= min.angle),
mapping = aes(x = x, y = y, label = name, angle = rotation, size = font.size)) +
scale_size_identity() # Ensure the sizes are used as is
# add theme
if (!is.null(theme.adjust)) sunb = sunb + theme.adjust
return(sunb)
}
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
deg_enrichment_df$log_pvalue <- -log10(as.numeric(deg_enrichment_df$old_specific_up_deg))
title <- "g50_old_specific_up_deg"
mdf$DEG_log_pvalue <- unlist(sapply(rownames(mdf), function(x) {
if(x %in% deg_enrichment_df$module) {
return(deg_enrichment_df[deg_enrichment_df$module == x, "log_pvalue"])
} else {
return(1) # 값이 없을 경우 기본값으로 1 사용
}}))
#EE2E31
#004F98
cutoffs <- c(0, -log10(1e-125), -log10(1e-30)) # legend 색조정 파트
cutoffs_norm <- (cutoffs - min(cutoffs)) / (max(cutoffs) - min(cutoffs))
colors <- c("white", "#EE2E31", "#EE2E31")
sbobj = draw_sunburst_wt_fill(module.df = mdf,feat.col = "DEG_log_pvalue",log.transform = FALSE,
fill.type = "continuous",
fill.scale = scale_fill_gradientn(colors = colors, values = cutoffs_norm, na.value = "white"),
id.col = "module.id",parent.col = "module.parent") + ggtitle(title) +
theme(panel.background = element_rect(fill = "white", color = NA),
plot.background = element_rect(fill = "white", color = NA))
ggsave("Oldsunburst.png", sbobj, width = 8)
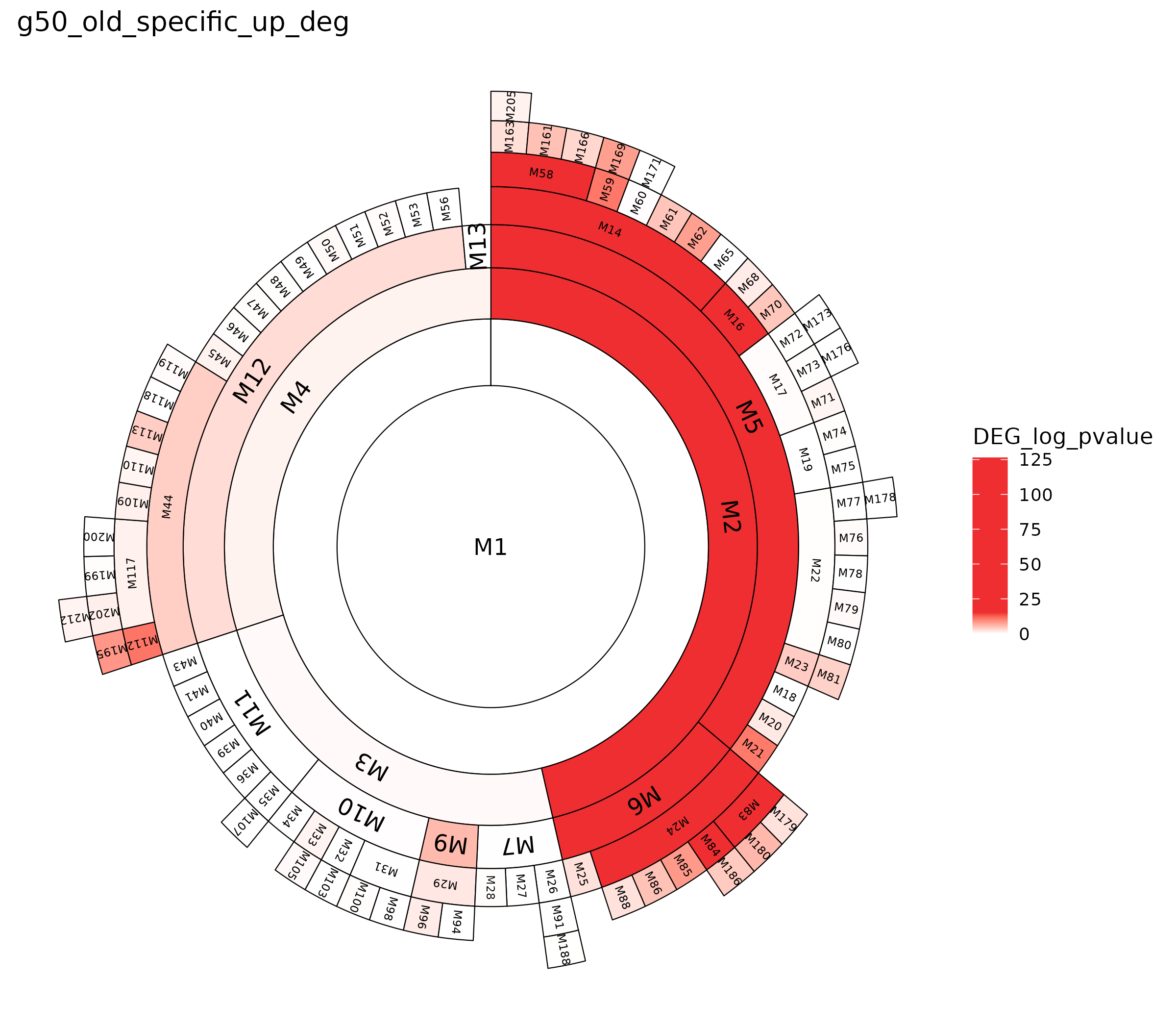
Enriched module -> GO functional annotation
Module 따로 떼서 GO term돌려보기 (not GSEA)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
library(clusterProfiler)
library(AnnotationDbi)
library(org.Hs.eg.db)
library(fgsea)
library(presto)
library(tibble)
library(msigdbr)
output.summary <- readRDS("../Orig/MEGENA_Mono_wSC_wo0_g50/MEGENA.summary.RDS")
m <- output.summary$module.table
ModuleGenes <- m[rownames(m)=="c1_2","module.hub"]
ModuleGenes <- strsplit(ModuleGenes, ",")[[1]]
ModuleGenes <- gsub("\\(.*\\)", "", ModuleGenes)
gobp <- clusterProfiler::enrichGO(ModuleGenes,
"org.Hs.eg.db", keyType = "SYMBOL",ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,qvalueCutoff = 0.05)
enr_go <- clusterProfiler::simplify(gobp)
# Select the top 5 terms based on FDR
top5_terms <- as.data.frame(enr_go) %>% arrange(p.adjust) %>% head(5)
top5_terms$FDR <- -log10(top5_terms$p.adjust)
# Create the bar plot
bar_plot <- ggplot(top5_terms, aes(x = reorder(Description, FDR), y = FDR)) +
geom_bar(stat = "identity",fill = "#3d3d3d", width = 0.7) +
coord_flip() + # Flip coordinates for horizontal bars
labs(x = "GO Terms", y = "-log(FDR)", title = "Old: M2") + theme_light() +
theme(text = element_text(size = 16),
axis.title = element_text(size = 12), # Axis titles
axis.text = element_text(size = 14))
ggsave(filename = "MEGENAg50_M2_GO.pdf", plot = bar_plot, width = 7, height = 3, dpi = 300)
References
- https://rpubs.com/grrompala/656670
- https://github.com/songw01/AD_scRNAseq_companion/blob/master/scripts/Section_K_Gene_Network_Analysis.Rmd
- chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/ModulePreservation/Tutorials/MiniTutorial-MouseLiver.pdf
- https://cran.r-project.org/web/packages/MEGENA/vignettes/MEGENA_pipeline_10062016.html
- https://rpubs.com/grrompala/656670
This post is licensed under
CC BY 4.0
by the author.