공개 데이터 다운받아서 재분석 하는 방법
참고 할만한 페이지
기본 분석 툴: CellRanger, Seurat 사용 가정
Raw data(.fastq) 제공하는 경우
SRA에서 다운 받아서 진행할때..1
➯ SRA에서 데이터 다운 받는법
예시로 내가 받은 한 샘플을 가져옴!
“GSM4577080: cerebral organoid 1; Homo sapiens; RNA-Seq(SRR11867465)”
10x chromium 기계로 생산한 single cell RNA-seq data다. SRAtoolkit으로 다운받으면 이렇게 세개의 파일이 받아진다.
1
2
3
SRR11867465_1.fastq
SRR11867465_2.fastq
SRR11867465_3.fastq
SRA toolkit으로 다운받을때, 처음부터 original format으로 다운받는 방법은 없나?
업로드한 사람이 올린 파일명 그대로 다운받을 수 있으면 좋을텐데..
SRA Browser를 통해 metadata 확인하면,
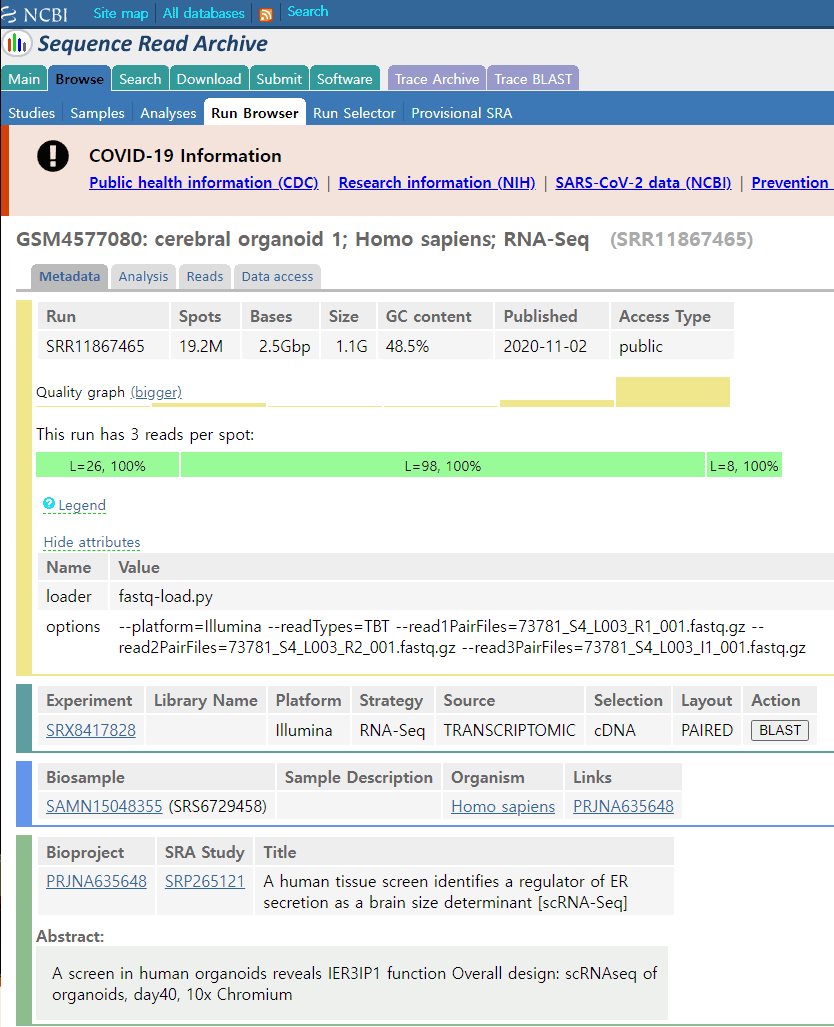
- 3개의 read의 길이는 26bp, 98bp, 8bp 임
➯ CellRanger 홈페이지에서 제공하는 정보를 바탕으로 이 데이터가 “Single Cell 3’ v2 (single indexed)” 로 만들어진 것을 알 수 있다. 또한 이 길이로 1,2,3.fastq가 순서대로 Read 1, Read 2, Index 1. - CellRanger는
bcl2fastq결과 파일 형식으로 된 fastq file을 받기 때문에 이름을 이에 맞게 바꿔줘야한다.
[Sample Name]S1_L00[Lane Number][Read Type]_001.fastq.gz
1
2
3
mv SRR11867465_1.fastq SRR11867465_S1_L001_R1_001.fastq
mv SRR11867465_2.fastq SRR11867465_S1_L001_R2_001.fastq
mv SRR11867465_3.fastq SRR11867465_S1_L001_I1_001.fastq
바꿨으면 input은 준비 끝!
Single-cell RNA-sequencing Analysis 여기 정리한대로 CellRanger count step부터 돌리면 된다.
Cellranger results(barcodes.tsv, gene.tsv, matrix.mtx) 제공하는 경우
기본적으로 cellrnager 이후 seurat object생성하는 방법과 동일하게 진행한다.
👉 SCP1038 single cell nucleus RNA seq dataset
- Download Mouse ileum all cells (10X) data and processed data(tSNE & clustering results)
msi.barcodes.tsv,msi.genes.tsv,gene_sorted-msi.matrix.mtx,msi.tsne2.txt
Change the file name. cellranger결과로 나온 것과 동일하게barcodes.tsv,genes.tsv,matrix.txt로 변경.
1
2
3
mv msi.barcodes.tsv barcodes.tsv
mv gene_sorted-msi.matrix.mtx matrix.mtx
mv msi.genes.tsv genes.tsv
- Make seurat object
1
2
3
4
5
6
SCP.data <- Read10X("/SCP_data_download_dir_location")
SCP <- CreateSeuratObject(counts = SCP.data, project = "SCP1038")
SCP
#An object of class Seurat
#22595 features across 79293 samples within 1 assay
#Active assay: RNA (22595 features, 0 variable features)
Error in '[.data.frame'(feature.names, , gene.column) :
Read10X 불러들여오는데 다음과 같이 error가 난다면 genes.tsv 파일을 열어서 column이 몇개인지 확인해보자. 한 column밖에 없다면 gene.column=1 옵션을 추가해주자.
1
2
3
SCP.data <- Read10X("/SCP_data_download_dir_location")
Error in `[.data.frame`(feature.names, , gene.column) :
undefined columns selected
1
2
3
4
5
6
7
8
9
10
11
$ head genes.tsv
0610007P14Rik
0610009B22Rik
0610009L18Rik
0610009O20Rik
0610010F05Rik
0610012D04Rik
0610012G03Rik
0610025J13Rik
0610030E20Rik
0610031O16Rik
- Load meta data
1
2
3
4
5
meta <- read.table("msi.tsne2.txt", sep='\t', header=TRUE, stringsAsFactors=FALSE)
meta$X <- as.numeric(meta$X)
meta$Y <- as.numeric(meta$Y)
meta$LABEL <- as.factor(meta$LABEL)
meta <- meta[-1,]
- Add meta data. 전에 만들어준 seurat object에 meta data(cluser annotation results)를 추가해준다.
1
SCP$label <- meta$LABEL
- 이후에 normalization부터 다시 분석 시작하면 됨.
Reference
- https://bioinformaticsworkbook.org/dataAnalysis/RNA-Seq/Single_Cell_RNAseq/Chromium_Cell_Ranger.html#gsc.tab=0
This post is licensed under CC BY 4.0 by the author.