SuperCell: sparsity 극복 시리즈 02
지난번에.. data sparsity를 해결하기 위해 MetaCell이라는 툴을 사용했었는데
👉 MetaCell R version: sparsity 극복법 01, pseudobulk 만들기 포스트 참조
내가 granularity value(얼마나 뭉칠것인지)를 잘못 설정한것인지 결과가 예상외로 별로여서 비슷한 컨셉의 다른 툴을 사용해봤다.
SuperCell
📁 Bilous, Mariia, et al. “Metacells untangle large and complex single-cell transcriptome networks.” BMC bioinformatics 23.1 (2022): 336.
MetaCell2가 나온시기와 비슷하게 2022년에 나왔다.
저번 포스팅에서도 그랬지만 이렇게 tool을 사용해서 계산된 pseudo-cell을 Metacell이라고 부르는듯. 제일 처음 이런 컨셉을 보인게 MetaCell논문이라서 계속 이렇게 부르는건가? 나도 앞으로 + 이 글에서, SuperCell 툴을 사용해서 만들었지만 그 결과로 얻은 pseudo-singlecell 을 Metacell이라고 부를 예정.
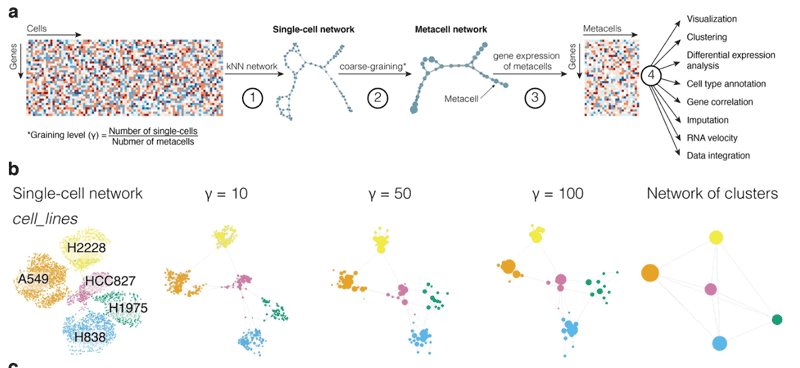
How it works
A single-cell network is computed based on cell-to-cell similarity (in transcriptomic space)
Highly similar cells are identified as those forming dense regions in the single-cell network and merged together into metacells (coarse-graining)
Transcriptomic information within each metacell is combined (average or sum).
Metacell data are used for the downstream analyses instead of large-scale single-cell data
Cell type같이 생물학적 개념을 포괄적으로 잡는 큰 cell 그룹을 식별하는 clustering과 달리, Metacell의 목적은 유사한 transcriptomic profile을 공유하고 반복적인 정보를 갖는 cell들은 하나로 합치는 것! 따라서 Metacell은 scRNA-seq data에서 중복된 정보를 제거하고 생물학적 다양성은 보존하는 compromise structure을 갖는다
Metacell구축에서 중요한 개념은 graining level (γ, G)로, the ratio between the number of single cells in the initial data and the number of metacells 로 정의된다. 저자는 G value를 10~50사이로 적용하길 권하고 있는데, 이 정도 수치가 downstream analysis하기에 resource도 아끼고 single-cell에서 찾았던 대부분의 결과를 보존하는 적당한 수치라고 함.
MetaCell과의 차이점
SuperCell:
- scRNA-seq 구조 단순화를 위해 coarse-graining framework 사용
- k-nearest neighbors (kNN) 알고리즘 사용해서, gene expression matrix에서 network 계산
- 각 singlecell의 Similarity 계산해서 -> 사용자 지정 graining level (g, gamma)의 MetaCell로 뭉쳐짐
- metacell의 gene expression matrix는 각 metacell에 포함된 single-cell들의 gene expression의 평균값으로 구성.
- 한번 metacell을 계산하면 single-cell analysis에서 할 수 있는 대부분의 downstream analysis 가능 (e.g. clustering, DEG, Cell type annotation etc.)
- SuperCell의 주된 목적은, 데이터 전체 구조는 보존하면서 복잡성을 줄이는데 있음
- 논문에서 보여주는 결과에 의하면 높은 수준으로 single-cell level에서 찾을 수 있는 clustering결과와 DEG결과를 metacell level에서도 확인 할 수 있었으며, metacell별로 purity를 계산할 수 있는데 거의 1에 가까운 수준으로 같은 종류의 celltype이 하나의 metacell로 뭉쳐짐
MetaCell:
- MetaCell은 graph partition approach를 사용함
- It defines metacells as groups of single-cell profiles that ideally represent resampling from the same cellular state.
- single-cell profiles간의 similarities를 계산해서 graph partition으로 metacell을 계산함.
- MetaCell툴의 목적은 signal-to-noise ratio를 높이는 것. over-smoothing 또는 잘못 계산된 modeling assumption으로부터 발생되는 bias를 계산에 포함시키지 않기 위해 노력한다
- large dataset에서도 효율적으로 작동하고, identify well-defined, discrete, and highly granular states.
SuperCell focuses on simplifying the data through a coarse-graining approach, creating metacells by merging densely connected cells. On the other hand, MetaCell defines metacells through a graph partition approach and emphasizes the unbiased characterization of cell types and expression gradients. Both tools aim to increase the efficiency and accuracy of analyzing complex single-cell datasets, but they do so through different methodologies and with a focus on different aspects of the data.
Installation
참고하면 좋을 페이지들
🔗 SuperCell github page
🔗 Workshop material: Simplifying large and complex single-cell RNA-Seq data with metacells
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
# dependencies
install.packages("igraph")
install.packages("RANN")
install.packages("WeightedCluster")
install.packages("corpcor")
install.packages("weights")
install.packages("Hmisc")
install.packages("Matrix")
install.packages("patchwork")
install.packages("plyr")
install.packages("irlba")
# install SuperCell
if (!requireNamespace("remotes")) install.packages("remotes")
remotes::install_github("GfellerLab/SuperCell")
# load package
library(SuperCell)
Finding Optimal G
👇 Trial01. avg. read depth & elbow point
Defining Function
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
makeSeuratMC <- function(GEX.seurOBJ, gamma= NULL,
genes = NULL,
metaFields = c("PatientID_re", "Condition", "SampleID_re", "Annot.L1", "Annot.L2"), # purity 확인하고 싶은 ident
returnMC = F) {
if(is.null(genes)) { # 어떤 feature 쓸건지 정하는 부분, 나는 Mitochondrial, Ribosomal genes은 feature에 넣고 싶지 않아서 제거
genes <- VariableFeatures(GEX.seurOBJ,assay="RNA")[!grepl("^MT|RPL|RPS", VariableFeatures(GEX.seurOBJ,assay="RNA"))]}
MC <- SCimplify(GetAssayData(GEX.seurOBJ,assay="RNA", layer="data"), # normalized gene expression matrix
n.pc = 10,
k.knn = 5, # number of nearest neighbors to build kNN network
gamma = gamma, # graining level
genes.use = genes)# will be the ones used for integration if input is seurat integrated data
MC$purity <- supercell_purity(clusters = GEX.seurOBJ$wsnn_res.0.8, # purity 계산하기 위한 ident들 넣어주기
supercell_membership = MC$membership)
for (m in metaFields) {
MC[[m]]<- supercell_assign(clusters = GEX.seurOBJ@meta.data[,m], # single-cell assigment to cell lines (clusters)
supercell_membership = MC$membership, # single-cell assignment to super-cells
method = "absolute")}
#GE <- supercell_GE(as.matrix(GetAssayData(GEX.seurOBJ,assay="RNA",slot = "data")),groups = MC$membership)
MC.counts <- supercell_GE(
ge = GetAssayData(GEX.seurOBJ, slot = "counts"),
mode = "sum", # summing counts instead of the default averaging
groups = MC$membership)
GE <- Seurat::LogNormalize(MC.counts, verbose = FALSE)
seuratMC <- supercell_2_Seurat(SC.GE = GE,MC,fields = c(metaFields,"purity"))
res <- seuratMC
if (returnMC) {res <- list(seuratMC = seuratMC,SC = MC)}
return(res)
}
Optimal G 계산
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
# 결과 저장할 data frame 선언
optimalG <- data.frame(g=c(2:200), avgReadDepth=0)
tic("Finding Optimal G")
for(g in optimalG$g){
print(g)
supercells_g <- makeSeuratMC(GEX.seurOBJ,gamma=g,returnMC = T)
saveRDS(supercells_g,paste0("supercells_sum_g",g,".rds"))
#supercells_g <- readRDS(paste0("supercells_g",g,".rds"))
seuratMC <- supercells_g$seuratMC
expression_matrix <- GetAssayData(seuratMC)
cell_read_counts <- colSums(expression_matrix)
average_read_depth <- mean(cell_read_counts)
optimalG[optimalG$g==g,'avgReadDepth'] <- average_read_depth
}
toc()
write.table(optimalG, "optimalG_sum.txt", sep='\t') # 결과 저장
#data <- read.table("optimalG_sum.txt", sep='\t')
Elbow point 찾고 plotting
elbow를 찾는 방법이 여러개 있겠지만, 나는 양끝을 잇는 선을 구하고 그 선에서 각 값까지의 거리가 가장 먼 지점을 찾는 방식을 선택함
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
# Function to calculate the perpendicular distance from a point to a line
perpendicular_distance <- function(x1, y1, x2, y2, x0, y0) {
num <- abs((y2 - y1) * x0 - (x2 - x1) * y0 + x2 * y1 - y2 * x1)
den <- sqrt((y2 - y1)^2 + (x2 - x1)^2)
return (num / den)
}
# Coordinates of the first and last points
x1 <- data$g[1]
y1 <- data$avgReadDepth[1]
x2 <- data$g[nrow(data)]
y2 <- data$avgReadDepth[nrow(data)]
# Calculating the perpendicular distances for each point
distances <- mapply(perpendicular_distance, x1, y1, x2, y2, data$g, data$avgReadDepth)
# Finding the index of the maximum distance
max_distance_index <- which.max(distances)
elbow_point_g <- data$g[max_distance_index]
# Plotting the data
p <- ggplot(data, aes(x=g, y=avgReadDepth)) +
geom_line() +
geom_vline(xintercept=elbow_point_g, linetype="dashed", color="red") +
theme_minimal() +
ggtitle('Average Read Depth vs g with Elbow Point') +
xlab('g') +
ylab('Average Read Depth')
ggsave("optimalG_plot.png",p)
print(paste('Elbow Point at g:', elbow_point_g))
Results
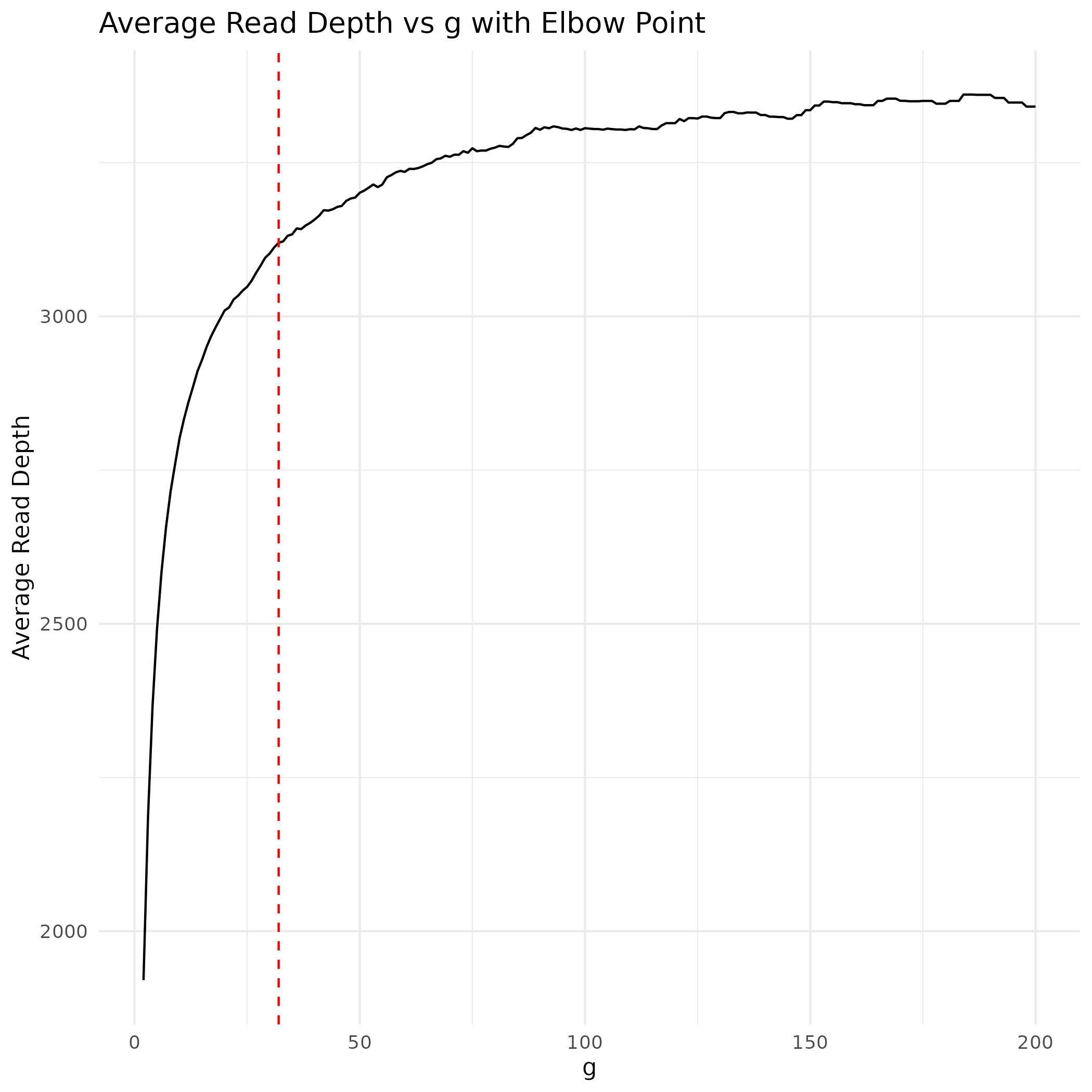
optimal g = 32
Insights
나쁘지 않은 방법인듯, 원래 SuperCell을 분석에 도입한 이유가 분석하고 있는 data가 pre-10X data라 cell당 발현값이 굉장히 낮아서 후속 분석을 하기 어려움이 있어서라 read depth의 saturation point를 찾아서 최대한 single-cell property를 보존하고 read depth가 최대에 가까운 g를 찾는!
근데 metacell gene expression matrix를 얻어서 MEGENA를 돌려봤는데 유의미한 gene network를 찾지 못함. 생가보다 g=32는 너무 많은 cell을 하나로 뭉그러뜨려 버리는 것 같아
Calculate metacell
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
library(Seurat)
library(SuperCell)
GEX <- readRDS("04.GEX_mapped.rds")
# SuperCell
sub <- get(paste0(cellType,"sub"))
sub = CreateSeuratObject(counts = LayerData(sub, assay = "RNA", layer = "counts"), project = "T", meta.data = sub@meta.data)
sub <- NormalizeData(sub)
sub <- FindVariableFeatures(sub,
selection.method = "disp", # "vst" is default
nfeatures = 1000)
hvg <- VariableFeatures(sub)
gamma <- 20 # Graining level
# Compute metacells using SuperCell package
MC <- SCimplify(
X = LayerData(sub, assay="RNA", layers="Data"), # single-cell log-normalized gene expression data
genes.use = hvg,
gamma = gamma,
n.pc = 10)
# Compute gene expression of metacells by simply averaging gene expression within each metacell
MC.ge <- supercell_GE(ge = LayerData(sub, assay="RNA", layers="Data"), groups = MC$membership)
##################>> MEGENA 돌리기
MC$purity <- supercell_purity(clusters = GEX$majorType[colnames(LayerData(GEX, assay="sketch", layers="counts"))],
supercell_membership = MC$membership)
metaFields = c("majorType") # 살리고 싶은 single cell label
for (m in metaFields) {
MC[[m]]<- supercell_assign(clusters = GEX@meta.data[colnames(LayerData(GEX, assay="sketch", layers="counts")),m], # single-cell assigment to cell lines (clusters)
supercell_membership = MC$membership, # single-cell assignment to super-cells
method = "absolute")
}
MC.counts <- supercell_GE(ge = LayerData(GEX, assay="sketch", layers="counts"),groups = MC$membership)
sobj <- CreateSeuratObject(counts = MC.counts)
sobj$majorType <- as.character(MC$majorType)
sobj <- NormalizeData(sobj)
sobj <- FindVariableFeatures(sobj, selection.method = "vst", nfeatures = 2000)
all.genes <- rownames(sobj)
sobj <- ScaleData(sobj, features = all.genes)
sobj <- RunPCA(sobj, features = VariableFeatures(object = sobj))
sobj <- FindNeighbors(sobj, dims = 1:10)
sobj <- FindClusters(sobj, resolution = 0.5)
sobj <- RunUMAP(sobj, dims = 1:10)
DimPlot(sobj, label = F , reduction = "umap",group.by="majorType",raster=FALSE)
ggsave("SC_umap_sketch_majorType.png",width=8)