Normalizing single cell RNA-seq data
Normaliztion이 필요한 이유 및 최신 툴
seurat normalizaion 과정 찾아보다가 보게 된 글. 정리용
Single cell RNA sequencing(scRNA-seq)의 목적은 대부분 sub-population을 찾고, DEG(Differential Gene Expression Analysis)분석 하는데에 있다. Curse of dimensionality 를 피하기 위해, cluster analysis할 때 Highly Variable Genes(HVG)를 사용한다. 여러 연구들에 따르면 HVG 뽑는게 raw counts matrices를 normalization하는 방법에 따라 영향을 받는다는 것이 드러났다.
:gem: 왜 normalization 해야 하는가?
Raw read counts로는 cell들간의 gene expression을 비교할 수 없다. (raw read counts에는 technical 및 ‘uninteresting’ biological variation까지 포함하고 있기 때문에,) 이런 Uninteresting biological variations를 제거하는(filter and regress) QC steps과 다른 방법들이 있다. PCR amplification bias는 Unique Molecular Identifiers(UMI)로 잘 보정되는 반면에, sequencing depth, cell lysis and reverse transcription efficiency의 차이 같은 다른 technical variations의 영향을 제거하기 위해서는 normalization이 필요하다.
Normalization process의 주된 목적은 true biological heterogeneity은 보존하면서 technical effects에 의한 영향을 제거하는 것이다. In a well normalized dataset, the variance of a gene should be independent of gene abundance and sequencing depth of a cell. ‘진짜’ DEG는 cell type에 걸쳐서 높은 분산을 보이는 반면, housekeeping genes은 낮은 분산을 보인다.
따라서 Normalization은, scRNA analysis의 후속분석에 큰 영향을 미치는 중요한 pre-processing step이다. Unfortunately, scRNA datasets은 일반적으로 bulkRNA seq method로 부터 내려오는 방법을 사용하여 normalized되는데, 이는 곧 다루겠지만, scRNA datasets의 technical variantions의 특성과 고유한 복잡성 때문에 부적절하다. In this blog post, scRNA-seq analysis과 관련하여 global scaling methods의 한계를 다루고, 최근 도입된 single cell analysis 특별 맞춤 normalization methods인 SCNorm과 SCTransform에 대해서 논의해 볼 것이다.
Global Scaling methods
Traditionally, raw expression counts across cells were normalized for sequencing depth with RPKM (Reads per Kilobase Million), FPKM (Fragments per Kilobase Million) or TPM (Transcripts per Million) methods. Normalization of bulk RNA seq datasets (RPKM, FPKM, TPM)
이런 방법들은 within-sample normalization으로는 효과적이지만, differential expression analysis in-between samples에는 적합하지 않다고 알려져 있다. 예를 들어, 두 유전자 A와 B의 발현을 두가지 condition - control과 treated에서 비교 하는 경우를 생각해보자. Gene A는 두 condition에서 같은 level로 발현됐고, 반면 gene B는 treated cell에서 2배 더 높게 발현됐다. TPM normalization은 absolute expression은 relative expression으로 전환하므로, Gene A가 differentially expressed 된다는 결론을 내릴 수 있다. 물론 이 효과는 단지 gene B와 비교한 결과일뿐이지만.
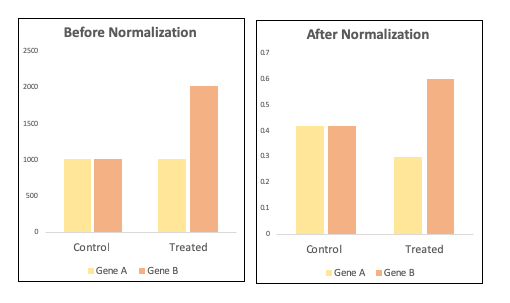 TPM normalization is unsuitable for differential expression analysis
TPM normalization is unsuitable for differential expression analysis
between-sample normalizations을 위해 개발된 다른 방법들도 있다.; TMM (trimmed mean of M-values) and DESeq이 가장 잘 알려져 있다. Both strategies follow the same motivation: to bring cell-specific measures onto a common scale by standardizing a quantity of interest across cells, while assuming that most genes are not differentially expressed.
non-differentially expressed genes을 바탕으로 이런 방법들에 의해 계산되는 the global scale factors는 각 sample마다 하나씩 적용된다. All genes in cells of a sample are then scaled using the same factor. 이 방법들은 bulk RNA-seq에서는 뛰어난 성능을 보이지만, single-cell setting에서는 zero-expression value의 양 때문에 제대로 발휘되지 못한다. 또한, 이 방법들은 기본적인 RNA 양이 sample내의 모든 세포에서 일정하고 모든 유전자에 대해 single scaling factor를 적용 가능하다는 것을 가정하고 시작한다. 그러므로 Global scaling factor based normalization strategies는 0인 값이 엄청 많고 highly heterogenous한 scRNA dataset에서는 저조한 성능을 보인다.
Gene group based methods
이런 global scaling approach의 고유한 문제를 해결하기 위해, 두가지 interesting normalization methods가 최근에 소개되었다 -SCnorm (2017) and SCTransform (Seurat package v3, 2019).
SCnorm
For every gene, SCnorm estimates the dependence(의존성) of gene expression on sequencing depth by a quantile regression. 그 다음 유사한 의존성을 갖는 genes을 묶고, 각 그룹에 대한 scale factor를 측정하기 위해 second quantile regression진행한다. 모든 gene group은 마침내 group specific scale factor를 사용해 sequencing depth에 대한 adjust를 마치고, normalized estimates of expression을 얻는다. (Every gene group is finally adjusted for sequencing depth using the group specific scale factor to produce normalized estimates of expression.)
아래 figure는 single cell dataset에서 count-depth relationship을 보여주는데, Panel A는 unnormalized 또는 raw expression counts. 명백하게(As is evident,), a global scale factor based method (panel B) 가 SCnorm in panel C와 비교했을 때 normalization이 잘 안된 것을 확인할 수 있다.
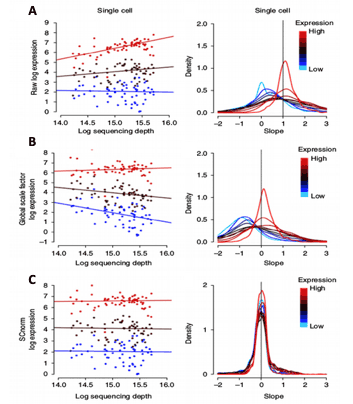 A: Raw Counts vs sequence depth, B: Global Scale Factor normalized vs sequence depth, C:SCnorm count vs sequence depth for 3 genes in a single cell dataset, edited from Bacher et al.
A: Raw Counts vs sequence depth, B: Global Scale Factor normalized vs sequence depth, C:SCnorm count vs sequence depth for 3 genes in a single cell dataset, edited from Bacher et al.
SCnorm: robust normalization of single-cell RNA-seq data
SCTransform
이 방법은 sequencing depth에 의한 variation을 제거하기 위해, regularized negative binomial model을 사용하여 UMI counts를 모델링한다. Briefly, 일단은 각 gene에 대해 generalized linear model (GLM)을 구축한다. 이때 sequencing depth를 independent variable로 UMI count를 response or dependent variable로 사용한다. The parameter estimates는 gene expression을 기반으로 regularized(or adjusted)된다. 두번째 negative binomial regression은 regularized parameters를 사용하여 적용된다. 이 model(residuals)의 결과는 the normalized expression levels for each gene 이다.
→모든 유전자를 normalize하기 위해 constant factor를 사용하기 보다, SCnorm과 SCTranscform methods 모두 gene-group specific factor를 학습해 사용했다. 이 factors은 각각 low, moderately, and highly expressed genes를 다루며, 진짜 biological heterogeneity는 보존하고 technical variation에 의한 영향만 제거한다.
Summary
normalization method를 선택하는 것은 HVG를 선택하고 이후 scRNA data의 downstream analyes에 큰 영향을 미친다. bulk RNA normalization methods를 scRNA dataset에 직접 적용하는 것은 부적절하고, SCNorm이나 SCTransform같은 pipeline으로 update할 필요가 있다.
- Reference
Normalizing single cell RNA sequencing data - Pitfalls and Recommendations