MuSiC: BulkRNA Deconvolution tool using scRNA-seq data
📑 Wang, Xuran, et al. “Bulk tissue cell type deconvolution with multi-subject single-cell expression reference.” Nature communications 10.1 (2019): 380.
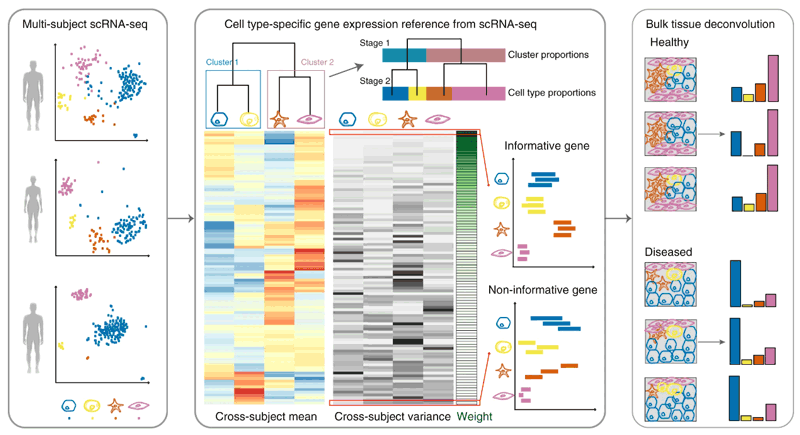
MUlti-Subject SIngle Cell deconvolution (MuSiC)
Deconvolution 이란..
Bulk RNA seq data 분석의 한 방법중 하나로. 모든 cell type이 구분없이 다 혼재되어있는 bulkRNA 데이터에서 여러 cell type의 비율을 추정하거나, 여러 샘플들에서 각 cell type의 유전자 발현 패턴을 분리해내기 위해 사용되는 분석 방법이다.
보통 Deconvolution 과정에는 2가지 input이 필요한데,..
-
- Reference profile
- 알아내고자하는 cell type의 순수한 유전자 발현 프로파일. 예전에는 FACs(Fluorescence-Activated Cell Sorting) 같은 걸로 분리된 세포군에서 얻어진 bulkRNA-seq data 또는 microarray data로 만들어진 database를 사용했다. 요즘은 single-cell data얻기가 더 쉬워져서 이를 활용한 툴이 많이 나오고 있는 추세다. like MuSiC!!
-
- Query profile (Mixed)
- cell type proportion을 알아내고자 하는 Bulk RNA-seq data. 여러 cell type이 혼합돼 있는 샘플에서 얻은 데이터.
이렇게 두가지 data를 갖고 deconvolution tool은 이 둘을 비교하면서 Query data내의 cell type 비율을 추정한다. 이 과정중에 어떤 방법론을 사용할 것이냐에 따라서 tool의 예측력같은게 차이나게 되는것. 보통 Linear regression, Machine learning, Matrix fractorization같은 방법을 사용한다.
MuSiC은..
wieghted non-negtive least squares regression(W-NNLS) 를 사용한 방법으로 linear regression의 일종을 사용한다. 간략히 설명하면 우리가 알아내고자 하는 cell type을 비율을 추정하는데 방정식을 사용하고 각 해가 음수가 아닐 것이라고 가정하고 이를 풀어가는 것이다. 잘 생각해 보면 당연한 것. cell type의 비율이 아무리 작아도 0이지 음수가 될 순 없으니께..
MuSic의 특장점?은 “marker gene consistency”라고 함. 지금까지 나온 CIBERSORT나 (CIBERSORx 아님) BSEQ-sc, TIMBER같은 툴들은 pre-selected cell type-specific marker gene을 사용해왔다면, MuSiC은 툴내에서 subject
지금까지의 툴들은 cross-subject heterogeneity와 with-in cell type stocahsticitiy를 무시하고 그냥 평균적으로 각 cell type의 marker로 사용되는 유전자들을 사용했다면, MuSic은 cross-subject and cross-cell consistency를 고려한 gene에 weight를 줘서 deconvolution한다.
무슨 말이냐면, 지금까지의 툴들은 single-cell data를 reference로 사용해서 bulkRNA를 deconvolution한다고 해도,.. 그냥 scRNA data에서 T cell cluster 특이적으로 logFC높은 유전자를 골라서 marker로 사용하거나 또는 실험적으로 많이 알려져 있는 surface protein marker를 기준으로 이들의 발현에 따라서 deconvolution을 진행했는데.. MuSiC이 꼬집는 이런 방법의 문제는 reference로 사용하는 데이터가 환자들의 phenotype에 따라서 같은 cell type이더라도 subject간의 발현 차이가 있을 수 있고(heterogeneity) 또는 같은 cell type내에서 stocahsticity에 의해서 어떤 유전자가 발현이 많거나/적게 캡쳐됐을 수 있는데 이런 것들을 고려하지 않고 따지자면 noise를 계산에 집어 넣었다는 것이다.
예를 들어 유전자 A가 T cell population의 marker로 잡혔는데 알고 봤더니 특정 질환을 앓는 환자의 Tcell에서 강력하게 발현되는 유전자였다. 이런 유전자들은 cross-subject consistency를 본다면 탈락. 비슷한 경우로 환자 phenotype으로 나눌 순 없지만 stocahsticity에 의해서 몇몇 cell에서 특정 유전자 B의 발현이 높게 나왔는데 population의 사이즈가 적어서 그만큼만으로도 B를 marker로 뽑히게 할 수 있었다면?..
MuSiC은 cross-subject and cross-cell consistency 를 유지하는 marker gene을 찾아내기 이전에, collinearity를 위해 tree-guided 단계를 추가했다. 별건 아니고 hierarchical clustering해서 얻을 수 있는 결과를 사용. 내가 input으로 주어진 모든 cell type에 대해서 clustering을 먼저 진해해서 이 cell type들을 더 큰 덩어리로 나누고 그 안에서 먼저 cluster-consistent genes을 찾고 큰 cluster별로 그 안에서 다시 intra-cell type variance를 보이는 유전자를 찾아서 deconvolution에 이용하게 된다. 만약 필요하다면 recursive하게 이 과정을 반복한다.
TL;DR
Subject간에서도 차이를 보이지 않고 constantly cell type을 대표하는 유전자들에 weight을 주어서 deconvolution에 사용하는 툴이다.
준비물
- Bulk RNA-seq data (Query) : raw count 사용 권장
- scRNA-seq data (Reference) : cell types는 이미 annotated 된 상태, raw UMI counts matrix
SingleCellExperimentobject를 input으로 받음 👉 Seurat v5 to SCE 포스트 참조
Installation
1
devtools::install_github('xuranw/MuSiC')
🚨 v.1.0.0 별 문제없이 설치 되긴했는데 돌리던 중 bug가 있는 것 같아서 music_prop() function 부분 코드 github에서 따서 개인적으로 수정후 사용. 아래내용 참조.
01. Data Preparations
앞서 말했듯이 MuSic은 single-cell data를 SCE 포맷으로 받기 때문에 변경해서 준비해준다.
1
2
3
4
5
6
7
8
GEX[["RNA"]] <- as(object = GEX[["RNA"]], Class = "Assay")
saveRDS(GEX,"GEX.v3.rds")
conda activate Seurat4
library(Seurat)
GEX <- readRDS("GEX.v3.rds")
test <- as.SingleCellExperiment(GEX)
saveRDS(test,"GEX.SCE.rds")
02. Estimation of cell type proportions
1
2
3
library(MuSiC)
library(reshape)
library(cowplot)
music_prop() function’s parameters
bulk.eset: ExpressionSet of bulk data; rownames = genename, colnames = sampleIDsc.eset: ExpressionSet of annotated single cell data;markers: vector or list of gene names, default as NULL. If NULL, then use all genes that overlapping in bulk and single-cell dataset;clusters: character, must be one of the column names of the phenoData from single cell data, used as clusters;samples: character, must be one of the column names of the phenoData from single cell data, used as samples;select.ct: vector of cell types included for deconvolution, default as NULL. If NULL, then use all cell types that provided by single-cell dataset.verbose: logical that toggles log messages.
bulk.eset(bulkRNA-seq expression matrix)는 값에 음수 들어있으면 나중에 에러난다.
논문에서도 raw count 값을 사용하는 것을 권장하고, TPM으로는 결과가 안나온다고 되어 있음. FPKM은 된다고 함.
시도해본 결과, CUF로는 돌아가지만 logCPM으로는 제대로 돌아가지 않는다.
bulk.eset에서 샘플별로 발현되는 유전자의 갯수와 이 유전자들이 single-cell data의 유전자 리스트와 겹치는지가 중요하다.
겹치는 유전자의 갯수가 20%이하면 에러나서 돌아가지 않는다.따라서 미리 input으로 주는 bulk.eset의 matrix를 정리해서 넣거나
(유전자 발현이 적은 샘플제거 및 single-cell dataset에서 발현되는 유전자만 row에 포함시키기)
markers옵션으로 사용할 유전자를 limit시키는것이 필요
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
# Bulk expression matrix
CUF = read.table("CUF.tsv",sep='\t',quote="",header=1)
# single-cell RNA-seq exp SCE obj.
sc.sce <- readRDS("GEX.SCE.rds")
# (optional) data cleaning
## rowname = hgnc_symbol로 하기 위해 정리
# bulk.mtx = CUF[CUF$hgnc_symbol!="",c(1,9:826)] # 818 samples
# gene_names <- bulk.mtx$hgnc_symbol
# bulk.mtx <- bulk.mtx %>% group_by(hgnc_symbol) %>%
# summarise(across(everything(), max, na.rm = TRUE)) %>% as.data.frame()
# rownames(bulk.mtx) <- bulk.mtx$hgnc_symbol
# bulk.mtx <- bulk.mtx[,-1]
## singlecell data와 일치하는 유전자만 사용
# bulk.mtx <- bulk.mtx[intersect(rownames(sc.sce),rownames(bulk.mtx)),]
## 발현되는 유전자의 갯수가 전체의 20% 이하인 샘플을 찾아서 제거
# threshold <- nrow(bulk.mtx) * 0.2
# bulk.mtx <- bulk.mtx[, colSums(bulk.mtx > 0) >= threshold] # 808 samples
# Estimate cell type proportions
Est.prop = music_prop(bulk.mtx, sc.sce, clusters = 'predicted.celltype.l2',samples = 'SampleID')
# saveRDS(Est.prop,"Est.prop_result.rds")
# names(Est.prop)
#[1] "Est.prop.weighted" "Est.prop.allgene" "Weight.gene" "r.squared.full" "Var.prop"
🚨 music_prop 수정된 코드
그냥 다운받은 library 그대로 돌렸더니, 충분한 양의 유전자가 발현되고 있음에도 불구하고 아래같은 에러메세지가 나오고 진행이 안돼서 몇줄 추가해서 수정함
1
2
3
4
5
6
7
8
9
# error code
Creating Relative Abudance Matrix...
Creating Variance Matrix...
Creating Library Size Matrix...
Used 13092 common genes...
Used 29 cell types in deconvolution...
X208.d0 has common genes 13063 ...
Error in music.iter(Yjg.temp, D1.temp, M.S, Sigma.temp, iter.max = iter.max, :
Not enough common genes!
👉👉👉 수정한 코드
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
## music_prop 코드 수정
music_prop = function(bulk.mtx, sc.sce, markers = NULL, clusters, samples, select.ct = NULL, cell_size = NULL, ct.cov = FALSE, verbose = TRUE,
iter.max = 1000, nu = 0.0001, eps = 0.01, centered = FALSE, normalize = FALSE, ... ){
bulk.gene = rownames(bulk.mtx)[rowMeans(bulk.mtx) != 0]
bulk.mtx = bulk.mtx[bulk.gene, ]
if(is.null(markers)){
sc.markers = bulk.gene
}else{
sc.markers = intersect(bulk.gene, unlist(markers))
}
sc.basis = music_basis(sc.sce, non.zero = TRUE, markers = sc.markers, clusters = clusters, samples = samples, select.ct = select.ct, cell_size = cell_size, ct.cov = ct.cov, verbose = verbose)
cm.gene = intersect( rownames(sc.basis$Disgn.mtx), bulk.gene )
if(is.null(markers)){
if(length(cm.gene)< 0.2*min(length(bulk.gene), nrow(sc.sce)) )
stop("Too few common genes!")
}else{
if(length(cm.gene)< 0.2*length(unlist(markers)))
stop("Too few common genes!")
}
if(verbose){message(paste('Used', length(cm.gene), 'common genes...'))}
m.sc = match(cm.gene, rownames(sc.basis$Disgn.mtx)); m.bulk = match(cm.gene, bulk.gene)
D1 = sc.basis$Disgn.mtx[m.sc, ];
M.S = colMeans(sc.basis$S, na.rm = T);
if(!is.null(cell_size)){
if(!is.data.frame(cell_size)){
stop("cell_size paramter should be a data.frame with 1st column for cell type names and 2nd column for cell sizes")
}else if(sum(names(M.S) %in% cell_size[, 1]) != length(names(M.S))){
stop("Cell type names in cell_size must match clusters")
}else if (any(is.na(as.numeric(cell_size[, 2])))){
stop("Cell sizes should all be numeric")
}
my_ms_names <- names(M.S)
cell_size <- cell_size[my_ms_names %in% cell_size[, 1], ]
M.S <- cell_size[match(my_ms_names, cell_size[, 1]),]
M.S <- M.S[, 2]
names(M.S) <- my_ms_names
}
Yjg = relative.ab(bulk.mtx[m.bulk, ]); N.bulk = ncol(bulk.mtx);
if(ct.cov){
Sigma.ct = sc.basis$Sigma.ct[, m.sc];
Est.prop.allgene = NULL
Est.prop.weighted = NULL
Weight.gene = NULL
r.squared.full = NULL
Var.prop = NULL
for(i in 1:N.bulk){
if(sum(Yjg[, i] == 0) > 0){
D1.temp = D1[Yjg[, i]!=0, ];
Yjg.temp = Yjg[Yjg[, i]!=0, i];
names(Yjg.temp) <- rownames(Yjg[Yjg[, i]!=0, ])
Sigma.ct.temp = Sigma.ct[, Yjg[,i]!=0];
if(verbose) message(paste(colnames(Yjg)[i], 'has common genes', sum(Yjg[, i] != 0), '...') )
}else{
D1.temp = D1;
Yjg.temp = Yjg[, i];
names(Yjg.temp) <- rownames(Yjg)
Sigma.ct.temp = Sigma.ct;
if(verbose) message(paste(colnames(Yjg)[i], 'has common genes', sum(Yjg[, i] != 0), '...'))
}
lm.D1.weighted = music.iter.ct(Yjg.temp, D1.temp, M.S, Sigma.ct.temp, iter.max = iter.max,
nu = nu, eps = eps, centered = centered, normalize = normalize)
Est.prop.allgene = rbind(Est.prop.allgene, lm.D1.weighted$p.nnls)
Est.prop.weighted = rbind(Est.prop.weighted, lm.D1.weighted$p.weight)
weight.gene.temp = rep(NA, nrow(Yjg)); weight.gene.temp[Yjg[,i]!=0] = lm.D1.weighted$weight.gene;
Weight.gene = cbind(Weight.gene, weight.gene.temp)
r.squared.full = c(r.squared.full, lm.D1.weighted$R.squared)
Var.prop = rbind(Var.prop, lm.D1.weighted$var.p)
}
}else{
Sigma = sc.basis$Sigma[m.sc, ];
valid.ct = (colSums(is.na(Sigma)) == 0)&(colSums(is.na(D1)) == 0)&(!is.na(M.S))
if(sum(valid.ct)<=1){
stop("Not enough valid cell type!")
}
if(verbose){message(paste('Used', sum(valid.ct), 'cell types in deconvolution...' ))}
D1 = D1[, valid.ct]; M.S = M.S[valid.ct]; Sigma = Sigma[, valid.ct];
Est.prop.allgene = NULL
Est.prop.weighted = NULL
Weight.gene = NULL
r.squared.full = NULL
Var.prop = NULL
for(i in 1:N.bulk){
if(sum(Yjg[, i] == 0) > 0){
D1.temp = D1[Yjg[, i]!=0, ];
Yjg.temp = Yjg[Yjg[, i]!=0, i];
names(Yjg.temp) <- rownames(Yjg[Yjg[, i]!=0, ])
Sigma.temp = Sigma[Yjg[,i]!=0, ];
if(verbose) message(paste(colnames(Yjg)[i], 'has common genes', sum(Yjg[, i] != 0), '...') )
}else{
D1.temp = D1;
Yjg.temp = Yjg[, i];
names(Yjg.temp) <- rownames(Yjg)
Sigma.temp = Sigma;
if(verbose) message(paste(colnames(Yjg)[i], 'has common genes', sum(Yjg[, i] != 0), '...'))
}
lm.D1.weighted = music.iter(Yjg.temp, D1.temp, M.S, Sigma.temp, iter.max = iter.max,
nu = nu, eps = eps, centered = centered, normalize = normalize)
Est.prop.allgene = rbind(Est.prop.allgene, lm.D1.weighted$p.nnls)
Est.prop.weighted = rbind(Est.prop.weighted, lm.D1.weighted$p.weight)
weight.gene.temp = rep(NA, nrow(Yjg)); weight.gene.temp[Yjg[,i]!=0] = lm.D1.weighted$weight.gene;
Weight.gene = cbind(Weight.gene, weight.gene.temp)
r.squared.full = c(r.squared.full, lm.D1.weighted$R.squared)
Var.prop = rbind(Var.prop, lm.D1.weighted$var.p)
}
}
colnames(Est.prop.weighted) = colnames(D1)
rownames(Est.prop.weighted) = colnames(Yjg)
colnames(Est.prop.allgene) = colnames(D1)
rownames(Est.prop.allgene) = colnames(Yjg)
names(r.squared.full) = colnames(Yjg)
colnames(Weight.gene) = colnames(Yjg)
rownames(Weight.gene) = cm.gene
colnames(Var.prop) = colnames(D1)
rownames(Var.prop) = colnames(Yjg)
return(list(Est.prop.weighted = Est.prop.weighted, Est.prop.allgene = Est.prop.allgene,
Weight.gene = Weight.gene, r.squared.full = r.squared.full, Var.prop = Var.prop))
}
🚨 music_prop.cluster 수정된 코드
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
#music_prop.cluster 코드 수정
music_prop.cluster = function(bulk.mtx, sc.sce, group.markers, groups, clusters, samples, clusters.type,
verbose = TRUE, iter.max = 1000, nu = 0.0001, eps = 0.01, centered = FALSE, normalize = FALSE, ... ){
bulk.gene = rownames(bulk.mtx)[rowMeans(bulk.mtx) != 0]
bulk.mtx = bulk.mtx[bulk.gene, ]
select.ct = unlist(clusters.type)
if(length(setdiff(names(group.markers), names(clusters.type))) > 0 || length(setdiff(names(clusters.type), names(group.markers))) > 0){
stop("Cluster number is not matching!")
}else{
group.markers = group.markers[names(clusters.type)]
}
if(verbose){message('Start: cluster estimations...')}
cluster.sc.basis = music_basis(sc.sce, non.zero = TRUE, markers = NULL, clusters = groups, samples = samples, select.ct = names(clusters.type), verbose = verbose)
if(verbose){message('Start: cell type estimations...')}
sc.basis = music_basis(sc.sce, non.zero = TRUE, markers = NULL, clusters = clusters, samples = samples, select.ct = select.ct, verbose = verbose)
cm.gene = intersect(rownames(sc.basis$Disgn.mtx), bulk.gene)
if(length(cm.gene)< 0.2*min(length(bulk.gene), nrow(sc.sce)) ){
stop("Too few common genes!")
}
if(verbose){message(paste('Used', length(cm.gene), 'common genes...'))}
m.sc = match(cm.gene, rownames(sc.basis$Disgn.mtx)); m.bulk = match(cm.gene, bulk.gene)
group.markers = lapply(group.markers, intersect, cm.gene)
D1 = sc.basis$Disgn.mtx[m.sc,]; M.S = sc.basis$M.S; Sigma = sc.basis$Sigma[m.sc, ]
cluster.select = setdiff(rownames(D1), unique(unlist(group.markers)))
cluster.diff = unique(unlist(group.markers))
D1.cluster = cluster.sc.basis$Disgn.mtx[cluster.select, ]; M.S.cluster = cluster.sc.basis$M.S;
Yjg = relative.ab(bulk.mtx[m.bulk, ]); N.bulk = ncol(bulk.mtx);
Sigma.cluster = cluster.sc.basis$Sigma[cluster.select, ];
D1.sub = cluster.sc.basis$Disgn.mtx[cluster.diff, ]; Sigma.sub = cluster.sc.basis$Sigma[cluster.diff, ];
Est.prop.weighted.cluster = NULL
for(i in 1:N.bulk){
if(sum(Yjg[, i] == 0) > 0){
name.temp = rownames(Yjg)[Yjg[, i] != 0]
D1.cluster.temp = D1.cluster[rownames(D1.cluster)%in%name.temp, ];
D1.sub.temp = D1.sub[rownames(D1.sub)%in%name.temp, ];
Yjg.temp = Yjg[Yjg[, i]!=0, i];
names(Yjg.temp) <- rownames(Yjg[Yjg[, i]!=0, ])
Sigma.cluster.temp = Sigma.cluster[rownames(Sigma.cluster)%in%name.temp, ];
Sigma.sub.temp = Sigma.sub[rownames(Sigma.sub)%in%name.temp, ];
if(verbose) message(paste(colnames(Yjg)[i], 'has common genes', sum(Yjg[, i] != 0), '...') )
}else{
D1.cluster.temp = D1.cluster;
D1.sub.temp = D1.sub;
Yjg.temp = Yjg[, i];
names(Yjg.temp) <- rownames(Yjg)
Sigma.cluster.temp = Sigma.cluster;
Sigma.sub.temp = Sigma.sub;
if(verbose) message(paste(colnames(Yjg)[i], 'has common genes', sum(Yjg[, i] != 0), '...'))
}
lm.D1.cluster = music.iter(Yjg.temp, D1.cluster.temp, M.S.cluster, Sigma.cluster.temp, iter.max = iter.max,
nu = nu, eps = eps, centered = centered, normalize = normalize)
p.weight = NULL
p.cluster.weight = lm.D1.cluster$p.weight
for(j in 1:length(clusters.type)){
if(length(clusters.type[[j]]) == 1){
p.weight = c(p.weight, p.cluster.weight[j])
}else{
if(p.cluster.weight[j] == 0){
p.weight = c(p.weight, rep(0, length(clusters.type[[j]])))
}else{
c.marker = intersect(group.markers[[j]], names(Yjg.temp))
Y.sub = D1.sub.temp[c.marker, j]*p.cluster.weight[j] +
(Yjg.temp[c.marker] - D1.sub.temp[c.marker, ]%*% p.cluster.weight) * p.cluster.weight[j]
names(Y.sub) = c.marker
Y.sub = Y.sub[Y.sub > 0]
lm.D1.sub = music.iter(Y.sub, D1[c.marker, clusters.type[[j]]], M.S[clusters.type[[j]]],
Sigma[c.marker, clusters.type[[j]]])
p.weight = c(p.weight, p.cluster.weight[j] * lm.D1.sub$p.weight)
}
}
}
Est.prop.weighted.cluster = rbind(Est.prop.weighted.cluster, p.weight)
}
colnames(Est.prop.weighted.cluster) = unlist(clusters.type)
rownames(Est.prop.weighted.cluster) = colnames(Yjg)
return(list(Est.prop.weighted.cluster = Est.prop.weighted.cluster))
}
Results
그래서 돌리고 난 Est.prop에는 샘플별로 예측된 결과가 저장되는데..
Est.prop.weighted: data.frame of MuSiC estimated proportions, subjects by cell types;Est.prop.allgene: data.frame of NNLS estimated proportions, subjects by cell types;Weight.gene: matrix, MuSiC estimated weight for each gene, genes by subjects;r.squared.full: vector of R squared from MuSiC estimated proportions for each subject;Var.prop: matrix of variance of MuSiC estimates.
NNLS란…
Non-Negative Least Squares의 약어. 비음수 최소제곱법이라고도함.
논문에선 CIBERSORT, BSEQ-sc와 함께 MuSiC의 결과를 평가하기 위한 방법중 하나로 사용함.linear regression을 위한 최적화 알고리즘 중 하나.
이 방법의 특징은 모든 x가 비음수여야 한다는 제약조건이 있음.여러 cell type의 혼합물 (bulkRNA-seq data)에서 각 cell type의 비율은 선형방정식으로 표현이 가능할텐데, 이때 각 cell type의 비율은 최소 0이지 음수가 될 수 없기 때문에
1
2
Est.prop$Est.prop.weighted # sample X celltypes
rowSums(Est.prop$Est.prop.weighted) # 전부 1
그림으로 표현
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
# 결과정리
m.prop = rbind(meltnew(Est.prop$Est.prop.weighted), meltnew(Est.prop$Est.prop.allgene))
colnames(m.prop) = c('Sub', 'CellType', 'Prop')
m.prop$CellType = factor(m.prop$CellType, levels = unique(sort(colData(sc.sce)$predicted.celltype.l2)))
m.prop$Method = factor(rep(c('MuSiC', 'NNLS'), each = ncol(expression_matrix_filtered)*length(levels(m.prop$CellType))), levels = c('MuSiC', 'NNLS'))
m.prop$day = sapply(m.prop$Sub,function(x) strsplit(as.character(x),split="[.]")[[1]][2])
m.prop$day = factor(m.prop$day,levels=c('d0','d3','d7','d28'))
m.prop$PatientID = sapply(m.prop$Sub,function(x) strsplit(as.character(x),split="[.]")[[1]][1])
# celltype별로 그림
cell_types <- unique(m.prop$CellType)
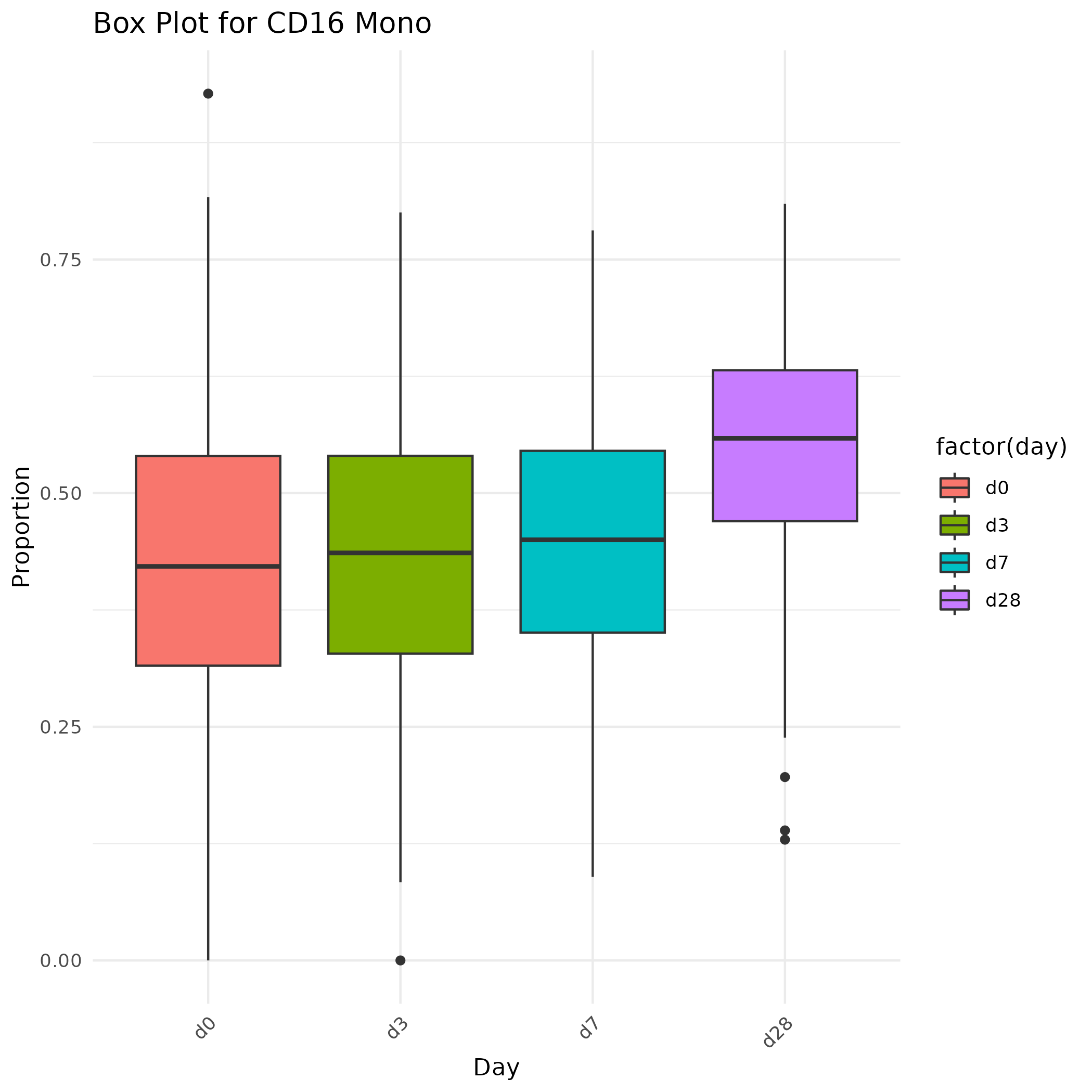
for (cell_type in cell_types) {
# 해당 Cell Type에 대한 데이터 추출
cell_type_data <- m.prop[m.prop$Method=="MuSiC",] %>% filter(CellType == cell_type)
# Box Plot 그리기
p <- ggplot(data = cell_type_data, aes(x = factor(day), y = Prop, fill = factor(day))) +
geom_boxplot() +
labs(title = paste("Box Plot for", cell_type), x = "Day", y = "Proportion") +
theme_minimal() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
# 그림 저장
ggsave(filename = paste(cell_type, "_MuSiC.png", sep = ""), plot = p)
}
이 툴 말고도 여러가지 다른 deconvolution 툴이 존재함.
끝
Reference
- https://xuranw.github.io/MuSiC/articles/pages/MuSiC2.html#sample-analysis