MetaCell R version: sparsity 극복법 01, pseudobulk 만들기
single data 분석이 항상 장점만 있는것은 아니다. 넘어야할 문제점들 또한 존재하는데 data sparsity 또한 그중 하나이다.
Data Sparsity란..
Data sparsity는 ‘cell에서 항상 모든 유전자가 발현되진 않는다’, ‘대부분의 유전자들은 아주 적은양만 발현되고 세포조절이 필요한 경우만 특별한 몇몇 유전자들의 발현이 높아지고, 조절이후에는 바로 원래상태로 돌아온다’ 같은 사실에 기반한다.
따라서 single cell sequencing의 결과인 count matrix를 놓고 봤을때 데이터 값이 대부분 0 또는 굉장히 낮은 값으로 채워져있는데, 이런 현상을 Data sparsity라고 한다. 0이 많은 데이터는 zero-inflated data라고 표현하기도함.
Data가 sparse했을때 여러 문제가 발생하는데,
- Comupational
- 데이터에 알맹이가 없이 메모리만 많이 차지해서, 계산시간을 늘림. 기존의 statistical method들은 이런 sparse/zero-inflated data처리에는 적합하지 않아 이를 위해 고안된 method사용해야함.
- Noise handling
- data가 너무 sparse하면 noise와 true biological signal을 구분하기 어려움.
- Feature selection
- 발현되는 유전자가 너무 적으면 feature slecetion에 문제가 생김. function에서 제공하는 feature의 기본 갯수는 3000개인데, 전체 cell에서 발현하는 평균유전자의 갯수가 이보다 적다면 informative한 정도로 줄여주는 조정과정이 필요함.
- Imputation
- Data sparsity를 해결하기 위한 다른 방법으로 imputation도 있음. 말그대로 값을 예측해서 채워주는 기법. 먼저 알고있는 유전자 발현을 기준으로 cell을 clustering한 뒤에, 같이 묶인 cell들의 발현량을 바탕으로 빈 부분의 count를 예측해서 채운뒤 후속 분석 하는것. 물론 가정은 ‘원래는 발현이 되고 있는 유전자였으나, 그 양이 매우적어 sequencing에는 잡히지 않았다.’ 실제 값이 아닌 예측한 값이므로 이런 방법을 사용하는 것에 대해서 각자 의견이 나뉨. standard방법은 아니다. 물론 오늘 소개할 metacell 같은 것도 standard는 아님ㅋㅋㅋ
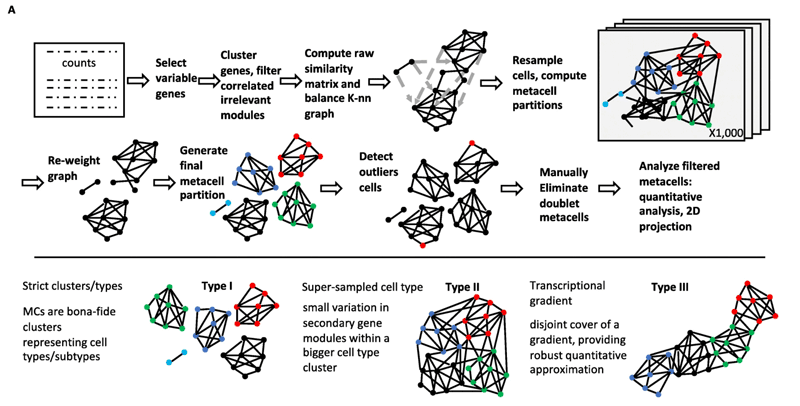
MetaCell
그래서 오늘 소개할 metacell은 data sparsity를 해결하기 위한 하나의 방법으로..
간단하게 이야기하면 비슷한 cell을 여러개 모아서 그들의 대표하는 특징을 갖는 가상의 세포(MetaCell)를 만들어냄으로서 data sparsity를 해결한다.
MetaCell papers
📁 Baran, Yael, et al. “MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions.” Genome biology 20.1 (2019): 1-19. 📁 Ben-Kiki, Oren, et al. “Metacell-2: a divide-and-conquer metacell algorithm for scalable scRNA-seq analysis.” Genome biology 23.1 (2022): 1-18.
MetaCell: R ver.
MetaCell2: python ver.
원리
오늘은 MetaCell R version 사용법 정리해보려고함. 이미 QC 끝난 Seurat obj로 시작할 예정.
아무래도 R, python 왔다갔다하는것보단 한 언어로 끝내는게 코드도 깔끔하니께
Installation
1
2
if (!require("BiocManager")) install.packages('BiocManager')
BiocManager::install("tanaylab/metacell")
Usage
기본적으로 seurat obj를 input으로 해서 metacell얻는 것까지 해보려함. MetaCell usage1 참고함.
01. Set up & Load input data
MetaCell 시작하려면 과정중에 나오는 object저장할 디렉토리(데이터베이스)가 필요하다. 여기선 testdb라는 이름으로 디렉토리를 만들고 package와 연결시켜준다. scdb_init function사용.
force_reinit=T 사용하면, 처음부터 다시시작. 기존에 testdb에 뭐가 들어있어도 무시하고 새로 덮어써버린다. Default 값은 FALSE, 원래 있던 데이터를 불러와서 사용하고 initiate하는 시간을 단축시킬수 있다. (optional)
testdb와 비슷하게 분석중에 나오는 figure를 저장할 디렉토리(데이터베이스)도 만들어서 package와 연결시켜주자. scfigs_init. 여기선 figs라는 이름으로 생성
1
2
3
4
5
6
7
library("metacell")
if(!dir.exists("testdb")) dir.create("testdb/")
scdb_init("testdb/", force_reinit=T)
if(!dir.exists("figs")) dir.create("figs/")
scfigs_init("figs/")
MetaCell은 10X 결과부터 시작할수도, SingleCellExperiment(sce) object로 부터 시작할수도 있다.
👇 10X 결과부터 시작하기
- obj_name : 아까만든 scdb에 저장할 이름. 앞으로 계속 사용할거니까 프로젝트명 같은걸로 네이밍하자
- base_dir : 10x결과 들어있는 위치
그외에, matrix_fn,genes_fn,cells_fn 같은 옵션이 있고 각 matrix, features, cellid file name을 지정할 수 있다.
1
2
mcell_import_scmat_10x("obj_name", base_dir="10x_results/outs/")
mat = scdb_mat("obj_name") # scdb에서 matrix가져오기
앞서 말했듯 나는 Seurat obj.부터 시작할거라, 먼저 seurat obj. ➯ sce obj.변환후 ➯ MetaCell로 import ➯ scdb에 추가2
원래 있던 Seurat obj.가 version 5 라면,..
👉 Seurat version5 다른 포맷으로 변환 SingleCellExperiment, loom, h5ad 포스팅참조
1
2
3
4
5
6
7
8
9
10
library(Seurat)
library(SeuratDisk)
Seurat.obj <- readRDS("OrigSeurat_v3.rds")
sce.obj <- as.SingleCellExperiment(Seurat.obj)
mat = scm_import_sce_to_mat(sce)
scdb_add_mat('obj_name', mat) # scdb에 추가, obj_name은 앞으로 계속 사용할거니 프로젝트명 같은걸로 정하기
# 하면 mat.gex.Rda 생김
02. Pre-processing
Exploring and filtering the UMI matrix
데이터 퀄리티 확인하고 low-quality gene, cell을 필터링해보자.
- distribution of UMI count per cell plot 그리기 (thresholded at 800 UMIs by default)
1
mcell_plot_umis_per_cell("obj_name",min_umis_cutoff=2^8.5)
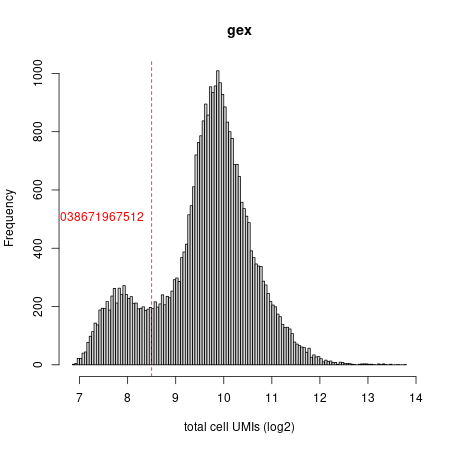
figs폴더아래에 obj_name.total_umi_distr.png 파일생김
Filtering uninformative genes
cell stress지수를 보이는 Mitochondrial gene이나 IG시리즈 유전자들은 feature selection에서 배제하기 위해 먼저 걸러낸다. mcell_mat_ignore_genes() 원래 있던 mat.obj_name.Rda 에 덮어씌워지는듯
본인 데이터에 맞게 어떤 유전자를 추가로 제외할지, 아니면 여기서는 제했어도 살려서 가져가던지 whatever you want 알아서 조절
1
2
3
4
5
6
7
8
9
10
mat = scdb_mat("obj_name")
nms = c(rownames(mat@mat), rownames(mat@ignore_gmat))
ig_genes = c(grep("^IGJ", nms, v=T),
grep("^IGH",nms,v=T),
grep("^IGK", nms, v=T),
grep("^IGL", nms, v=T))
bad_genes = unique(c(grep("^MT-", nms, v=T), grep("^MTMR", nms, v=T), grep("^MTND", nms, v=T),"NEAT1","TMSB4X", "TMSB10", ig_genes))
mcell_mat_ignore_genes(new_mat_id="obj_name", mat_id="obj_name", bad_genes, reverse=F)
Filtering low-expressed cells
유전자와 마찬가지로 쓸데없는, 전체적인 발현이 낮은, empty droplet일 가능성이 높은 cell도 후속 분석에서 제외할 수 있다.
나는 seurat obj 만들때 이미 low-quality cell은 다 제거했기 때문에 스킵
1
mcell_mat_ignore_small_cells("obj_name", "obj_name", 800)
Selecting feature genes
metacell graph를 만들기에 앞서 feature selection을 진행한다. var.mean과 cov값을 조정해서 몇개의 유전자를 feature로 사용할지 결정한다.
!!!한번에 결정하는 법아는 사람 댓글 좀 부탁드려요!!!
- variance mean(T_vm) : 0.08이 default인듯, 이 이상의 값을 넣으면
- T_tot: 전체 샘플에서 total umi count값이 200이상인 유전자만 사용 (왠만해서는 T_vm값으로 갯수 결정되는 듯, 이 값은 최악거르기느낌, 내 데이터에선 feature결과엔 별로 영향안주더라)
나는 visualization 까지 다 돌려보고 clustering결과 보고 조정해가면서 여러번 돌림. 너무 뭉쳐있으면 var.mean ⇈
1
2
3
4
mcell_add_gene_stat(gstat_id="obj_name", mat_id="obj_name", force=T) # gstat.gex.Rda
mcell_gset_filter_varmean(gset_id="obj_name_feats", gstat_id="obj_name", T_vm=0.3, force_new=T) # gset.gex_feats.Rda
mcell_gset_filter_cov(gset_id = "obj_name_feats", gstat_id="obj_name", T_tot=200, T_top3=2)
mcell_plot_gstats(gstat_id="obj_name", gset_id="obj_name_feats")
03. Run MetaCell
Building the balanced cell graph
첫번째 raw similarity matrix와 balanced KNN graph를 만든다.
1
2
3
4
5
mcell_add_cgraph_from_mat_bknn(mat_id="obj_name", # cgraph.gex_graph.Rda
gset_id = "obj_name_feats",
graph_id="obj_name_graph",
K=100,
dsamp=T)
여기서 K 값이 얼마나 돼야할지.. optimal K를 찾는건 따로 다루도록 하자.