[QC] DoubletFinder: doublet 제거법 01
Doublet이란?
Doublet은 두 개의 세포가 하나의 단일 세포로 인식돼서 분석할때 생기는 문제를 일컷는다. 이는 10X와 같이 droplet-based sequencing과정에서 주로 발생하는데.. 기계가 자동으로 cell suspension에서 단일 세포와 barcode정보를 담고 있는 bead를 하나씩 하나의 droplet에 담는 과정에서 technical error로 두개 이상의 세포가 하나의 droplet에 담기게 되면서 생성된다. 두개가 담기면 doublet, 더 많이 담기면 multiplet이라고 함.
 Chromium Next GEM Single Cell 3ʹ v3.1 manual
Chromium Next GEM Single Cell 3ʹ v3.1 manual
보통은 한 lane에 넣는 cell수가 많을수록 multiplet 발생 빈도가 올라간다고 한다.1 10X chromium 사에서는 각 loading cell number별로 발생할 수 있는 multiplet rate을 메뉴얼에 공개하고 있다2.
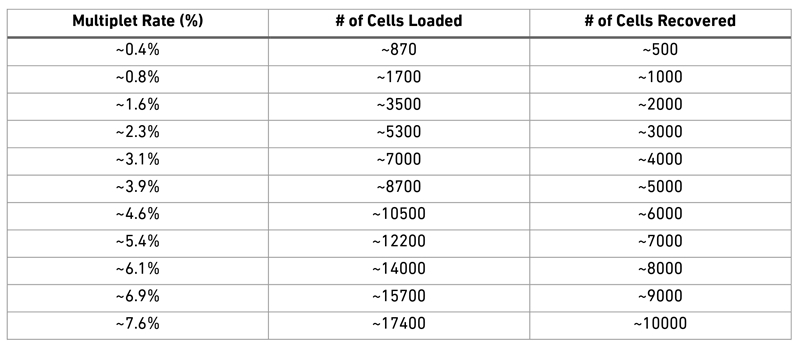 Importance of Loading Cell Concentration
Importance of Loading Cell Concentration
해결방법
Seurat 기본 메뉴얼에서는, 이상할정도로 높은 gene counts(nFeature_RNA)를 가진 cell을 dublet이나 multiplet으로 간주하고 제거하라고 이야기하고 있다3.
Outlier 수준으로 평균의 2배이상의 nFeature_RNA 값을 갖는다면 의심할법도 한데, 이 기준이 굉장히 주관적이고 애매모호하지 않은가.. 어디까지 outlier로 할 것이며, cancer cell같은 세포들은 안에서 다양한 metabolism pathway가 활성화되어 있어서 보통 상태의 normal cell보다 몇배에 달하는 유전자 종류가 발현되고 있을 수 있다.
이런 문제들 때문에 지금까지 여러 doublet를 골라내는 tool들이 개발됐고, DoubletFinder뿐만 아니라 Scrublet, DoubletDecon 같은 tool도 있다.
DoubletFinder
🔗github page
🔗publication link / Published: April 03, 2019 / DOI: https://doi.org/10.1016/j.cels.2019.03.003
Mechanism & Processing step
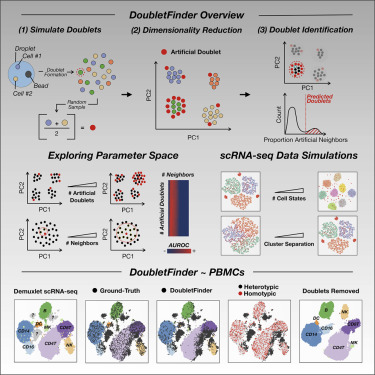
DoubletFinder의 작동방식은 다음과 같다.
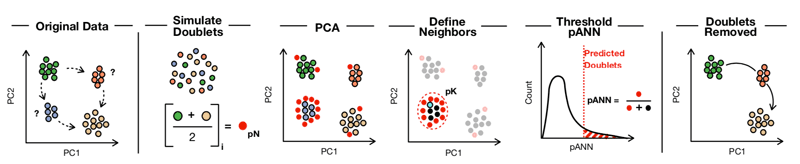
1. Generate artificial doublets from existing scRNA-seq data
DoubletFinder는 input으로 seurart object와 여러 parameter를 받는데, 이때 seurat object는 normalization과 clustering까지 마친 상태로 넣는다. DoubletFinder는 dataset의 값을 바탕으로 artificial doublet를 만들어낸다. 임의로 2개의 cluster(cell type)을 선택해 이들의 expression profile을 합쳐서 만듬. 만들어내는 artificial doublet의 수는 실제 데이터에 있는 doublet의 예상 비율을 기반으로 제작된다.
2. Pre-process merged real-artificial data
이렇게 만든 artificial doublets을 원래 데이터에 추가해서 pre-procssing을 진행. PCA로 PC distance matrix구하고 k-nearest neigbor(KNN) graph를 생성한다. Doublet은 보통 두가지 이상의 cell type을 섞어서 만든 것이기 때문에, cluster들의 사이에 위치함.
3. Rank order and threshold pANN values according to the expected number of doublets
전 단계에서 찾은 KNN graph 통해 각 cluster에서 artificial KNN의 비율(pANN)을 계산한다. 이 점수는 각 세포가 얼마나 doublet에 가까운지?/얼마나 doublet처럼 보이는지 나타낸다. 점수가 높을수록 doublet일 가능성이 높다.
4. Rank order and threshold pANN values according to the expected number of doublets
마지막으로 점수를 바탕으로 줄을 세우고 threshold를 결정해서 doublet과 singlet을 나눈다.
Input parameters
DoubletFinder는 기본 5가지의 input parameters를 요구한다.
-
- seu
- Seurat object, 이미 다 processed된 seurat obj여야함.
NormalizeData,FindVariableGenes,ScaleData,RunPCA,FindNeighbors,FindClusters,RunUMAP까지
-
- PCs
- 유의미한 PC range.
FindNeighbors등 돌릴때 사용했던 PC값 똑같이 사용하면 됨.
-
- pN
- 이 값에 의해 만들어질 aritificial doublets의 갯수가 정해진다. step2에서 만들어지는 real-artificial dataset 에서의 비율로 표현되는데, Default는 25%. 메뉴얼에 따르면 DoubletFinder의 성능은 pN값과는 무관하다고함. 25%보다 크거나 작아도 결과로 예측되는 doublet list는 동일하다는 의미
-
- pK
- pANN을 계산다는데 사용되는 PC neighborhood의 크기. dataset마다 pK값을 찾아서 정해햐하기 때문에 default가 제공되지 않는다. Optimal pK를 찾는 방법은 아래에서..
-
- nExp
- 마지막으로 doublet/singlet을 예측할때 pANN의 threshold를 정할때 사용함. 사용하는 platform별로 메뉴얼에서 제공하는 loading cell number 대비 예상 doublet 생성 비율을 참고해서 정하면 됨. (10x 예상 doublet 비율을 위에 표로 첨부)
Installation
1
remotes::install_github('chris-mcginnis-ucsf/DoubletFinder')
Usage
주의사항
sample별로 따로 돌려야함. 한 sample에 한번씩 돌리는걸로 생각하면 쉬움, 한 sample을 여러 lane으로 나눠서 돌리고 aggregate한 뒤에 DoubletFinder input으로 넣는건 괜춘. control, treated따로 인데 합쳐서 돌리는건 안됨.
Doublet말고 다른 low quality cell은 최대한 제거한 상태로 돌리자. 다음과 같은 특징을 가진 cluster 다 제거한 이후에 DoubletFinder진행. low RNA UMIs, High % mitochondrial reads, and/or Uninformative marker genes
pK Selection
앞서 말했듯이 pN값은 DoubletFinder의 성능에 별 영향을 주지 않는다.
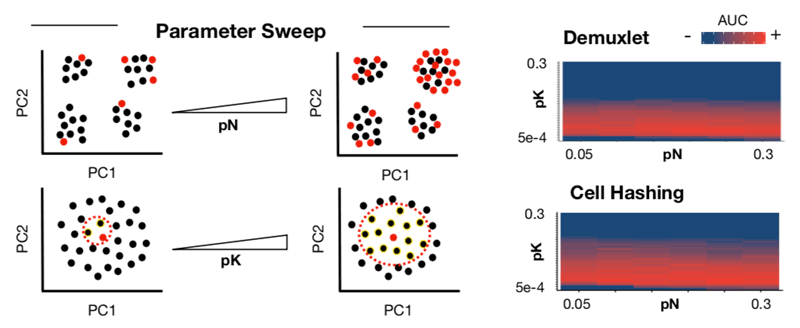
다양한 pN-pK parameter조합으로 ROC 분석을 진행해본 결과,
- Optimal pK은 cell state 갯수에 의해 결정된다.
- DoubletFinde의 성능은 transcriptonally-homogeneous한 데이터에서 최악이다.
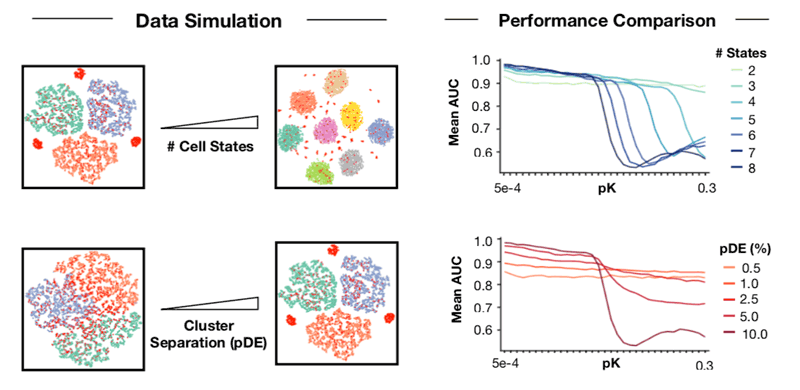
그래서 cell state 갯수는 dataset마다 다르기 때문에, DoubletFinder에서는 데이터에 맞는 optimal pK 값을 찾는 pK selection과정이 필수적이다. ground truth를 알고 있는 경우, (어떤 cell이 doublet인지 알고 있는 경우, 일부러 doublet을 생성해서 만든 샘플들,) parameter optimization하기 쉬웠을테지만 보통은 모르니까.. DoubletFinder에서는 이를 해결하기 위해, ground truth에 구애받지 않아며 AUC를 최대화하는 pK 값을 찾는 방법을 고안했고
Mean-variance normalized bimodality coefficient (BCmvn) 값을 활용하는 방법이다. BCmvn은 단일계수만으로 AUC를 최대화하는 pK 값을 결정할 수 있게 도와준다.
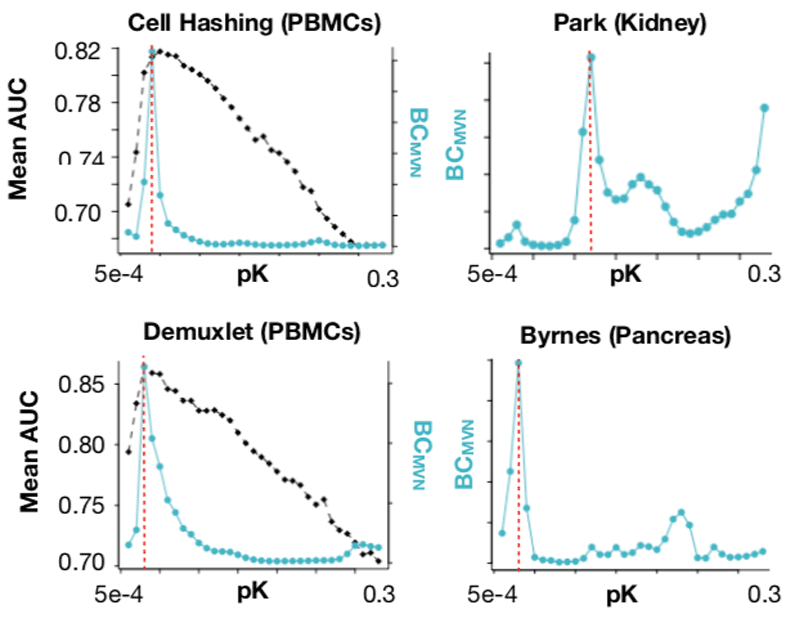
➥ BCmvn Peak가 보이는 pK값을 선택하면 됨. 아래 코드에선 최대 BCmvn값을 갖는 pK값을 선택하도록 자동화.
Code
Seurat version 5는 아직 지원되지 않는듯. 2023 Jul 기준.
만약 Seurat obj가 v.5 기준으로 생셩됐다면 downgraded 후에 진행하자.
seu[["RNA"]] <- as(object = seu[["RNA"]], Class = "Assay")
2024 이제 Seurat version 5 지원됨
- ground turth 있는 버전 없는 버전 둘다 포함하고 있으니까 맘대로 골라쓰쟈
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
#여러 샘플이라 가정
S1 <- readRDS('00.S1_raw.rds')
S2 <- readRDS('00.S2_raw.rds')
S3 <- readRDS('00.S3_raw.rds')
SampleList <- c('S1','S2','S3')
for(i in SampleList){
## Pre-process Seurat object (sctransform) -----------------------------------------------------------------------------------
# 어떤 방법으로 pre-processing할지는 자기맘.
seu <- get(i)
seu[["RNA"]] <- as(object = seu[["RNA"]], Class = "Assay")
seu <- NormalizeData(seu)
seu <- FindVariableFeatures(seu, selection.method = "vst", nfeatures = 2000)
seu <- ScaleData(seu)
seu <- RunPCA(seu)
seu <- RunUMAP(seu, dims = 1:10) # PC 몇개쓸진 자기맘
seu <- FindNeighbors(seu, dims=1:10)
seu <- FindClusters(seu)
## pK Identification (no ground-truth) ---------------------------------------------------------------------------------------
sweep.res.list <- paramSweep_v3(seu, PCs = 1:10, sct = FALSE)
sweep.stats <- summarizeSweep(sweep.res.list, GT = FALSE)
bcmvn <- find.pK(sweep.stats)
## pK Identification (ground-truth) ------------------------------------------------------------------------------------------
#sweep.res.list <- paramSweep_v3(seu, PCs = 1:10, sct = FALSE)
#seu$GT <- cell_species$species
#seu@meta.data[seu$GT%in%c("dog","human"),"GT"] <- 'Singlet'
#seu@meta.data[seu$GT=="mixed","GT"] <- 'Doublet'
#gt.calls <- seu@meta.data[rownames(sweep.res.list[[1]]), "GT"] ## GT is a vector containing "Singlet" and "Doublet" calls recorded using sample multiplexing classification and/or in silico geneotyping results
#sweep.stats <- summarizeSweep(sweep.res.list, GT = TRUE, GT.calls = gt.calls)
#bcmvn <- find.pK(sweep.stats)
ggplot(bcmvn, aes(pK, BCmetric, group=1)) + geom_point() + geom_line()
ggsave(paste0("DoubletFinder_",i,"_peak.png"))
pK <- bcmvn %>% filter(BCmetric==max(BCmetric)) %>% select(pK)
pK <- as.numeric(as.character(pK[[1]]))
print(paste0(i,":",pK))
## Homotypic Doublet Proportion Estimate -------------------------------------------------------------------------------------
annotations <- seu$seurat_clusters
multiRate= c('S1'=0.039,'S2'=0.054,'S3'=0.04) # 예상 doublet rate, 10X manual table참고
homotypic.prop <- modelHomotypic(annotations)
nExp_poi <- round(multiRate[i]*ncol(seu))
nExp_poi.adj <- round(nExp_poi*(1-homotypic.prop))
## Run DoubletFinder with varying classification stringencies ----------------------------------------------------------------
seu <- doubletFinder_v3(seu, PCs = 1:10, pN = 0.25, pK = pK, nExp = nExp_poi.adj, reuse.pANN = FALSE, sct = TRUE)
DF.name = colnames(seu@meta.data)[grepl("DF.classification", colnames(seu@meta.data))]
DimPlot(seu, group.by = DF.name) + NoAxes()
ggsave(paste0("DoubletFinder_",i,".png"))
VlnPlot(seu, features = "nFeature_RNA", group.by = DF.name, pt.size = 0.1)
ggsave(paste0("DoubletFinder_",i,"_vln.png"),width=14)
saveRDS(colnames(seu[, seu@meta.data[, DF.name] == "Singlet"]),paste0("DoubletFinder_",i,"_Singlet_cells.rds"))
saveRDS(colnames(seu[, seu@meta.data[, DF.name] == "Doublet"]),paste0("DoubletFinder_",i,"_Doublet_cells.rds"))
}
🚨 doubletFinder_v3 단계에서 에러
1
2
Error in vst(vst.flavor = "v2", umi = new("dgCMatrix", i = c(53L, 99L, :
Please install the glmGamPoi package. See https://github.com/const-ae/glmGamPoi for details.
해결방법: ‘glmGamPoi’설치
1
2
BiocManager::install("glmGamPoi")
conda install -c bioconda bioconductor-glmgampoi
🚨🚨 ‘glmGamPoi’ 설치중 dependency 에러
1
2
3
4
5
6
7
8
ERROR: dependency ‘Rhdf5lib’ is not available for package ‘rhdf5filters’
* removing ‘.../lib/R/library/rhdf5filters’
ERROR: dependencies ‘Rhdf5lib’, ‘rhdf5filters’ are not available for package ‘rhdf5’
* removing ‘.../lib/R/library/rhdf5’
ERROR: dependencies ‘rhdf5’, ‘rhdf5filters’, ‘Rhdf5lib’ are not available for package ‘HDF5Array’
* removing ‘.../lib/R/library/HDF5Array’
ERROR: dependency ‘HDF5Array’ is not available for package ‘glmGamPoi’
* removing ‘.../lib/R/library/glmGamPoi’
해결방법: ‘HDF5Array’설치
1
2
3
conda install -c bioconda bioconductor-hdf5array
conda install -c bioconda bioconductor-rhdf5
Results
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
# Doublet Finder 결과
cells <- c()
for(i in SampleList){
tmp <- readRDS(paste0("DoubletFinder_",i,"_Doublet_cells.rds"))
cells <- c(cells,paste0(i,"_",tmp))
}
#obj는 이미 integration마친 S1,S2,S3의 모음
obj$DoubletFinder <- "Singlet"
obj@meta.data[cells,"DoubletFinder"] <- "Doublet"
# table(subset(NYU,subset=DoubletFinder=='Doublet')$SampleID)
png("01.QC_firstUMAP_Doublet_DoubletFinder.png")
DimPlot(obj, reduction = "umap.unintegrated", group.by = "DoubletFinder",cols=c("red","grey"))
dev.off()
그림 그려보면 이렇게 doublet으로 예측된 cell들이 한 cluster에 뭉쳐있기도 하고, 군데군데 있기도함.
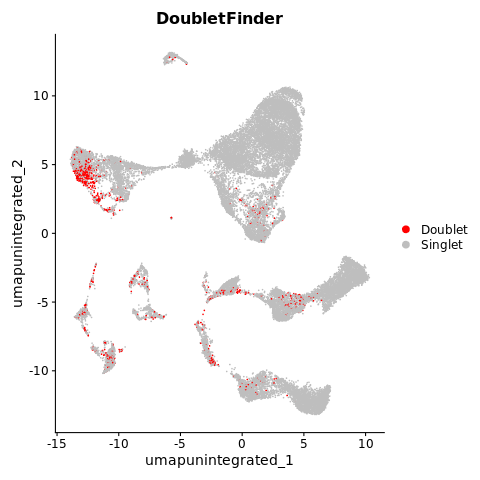
Cluster통으로 뺄지, doublet이라고 나온 애들만 빨지는 개인의 선택.
나는 이중에서도 nFeature가 Q3이상인 친구들만 빼는 방법을 선호함. Doublet은 두가지의 다른 타입의 셀이 합쳐졌다는 가정하에 정의되기 때문에.