[10X CellRanger] Multi pipeline, GEX/TCR/BCR/SP 한번에 처리하는법
요즘은 single-cell에서 gene expression만 보지 않고 한 cell에서도 surface protein expression level과 TCR, BCR clone도 함께 확인할 수 있다.
10X genomics에서도 이런 서비스를 제공하고 있으며, 이런 데이터 처리를 위해 CellRanger에 multi pipeline을 탑재해놨다. 보통 GEX만 처리하면 count를 쓸때 이런 multi-omic data처리하기 위해 multi 쓴다고 생각하면 됨.
참고하면 좋을 페이지
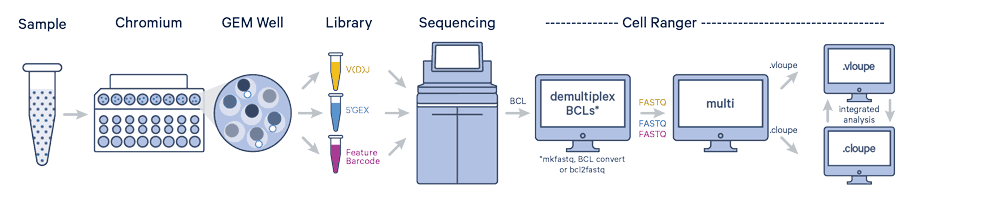 Overview of Multi-seq
Overview of Multi-seq
🎀Make fastq files from bcl
🍒Prerequisite
- bclfastq2 설치 👉여기 참고 (Cell Ranger (v7.1 and later)은 bcl2fastq v2.20이상 이 필요함)
- raw files
- barcode info.csv
raw files
.bcl file포함 아래같은 구조로 생겼다.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
├── Config
│ ├── Effective.cfg
│ ├── LaserPowerVariability.xml
│ ├── NovaSeqCalibration.cfg
│ ├── NovaSeqOverride.cfg
│ └── Options.cfg
├── CopyComplete.txt
├── Data
│ └── Intensities
├── InterOp
│ ├── AlignmentMetricsOut.bin
│ ├── BasecallingMetricsOut.bin
│ ├── C1.1
│ ├── ...
│ ├── C109.1
│ ├── CorrectedIntMetricsOut.bin
│ ├── EmpiricalPhasingMetricsOut.bin
│ ├── ErrorMetricsOut.bin
│ ├── EventMetricsOut.bin
│ ├── ExtendedTileMetricsOut.bin
│ ├── ExtractionMetricsOut.bin
│ ├── FWHMGridMetricsOut.bin
│ ├── ImageMetricsOut.bin
│ ├── OpticalModelMetricsOut.bin
│ ├── PFGridMetricsOut.bin
│ ├── QMetrics2030Out.bin
│ ├── QMetricsByLaneOut.bin
│ ├── QMetricsOut.bin
│ ├── RegistrationMetricsOut.bin
│ └── TileMetricsOut.bin
├── Logs
│ ├── 240516_A01097_0281_AHVKNTDMXY_Cycle0_Log.00.log
│ ├── ...
│ ├── 240516_A01097_0281_AHVKNTDMXY_Cycle99_Log.00.log
│ ├── 240516_A01097_0281_AHVKNTDMXY_Warnings_And_Errors_Log.00.log
│ ├── 240516_A01097_0281_AHVKNTDMXY_Wash.00.log
│ ├── 240516_A01097_0281_AHVKNTDMXY_Wash.01.log
│ ├── 240516_A01097_0281_AHVKNTDMXY_Wash.02.log
│ ├── A01097_2024-05-16__14_20_21_Side A_RunSetup.00.log
│ ├── A01097_2024-05-16_15-51-11_FM-AutoTilt_Report.csv
│ ├── A01097_2024-05-16_15-55-25_ObjectiveAirShieldPrerunPWMSweep_Test_Results.csv
│ ├── A01097_2024-05-16_18-47-30_FM-AutoTilt_Report.csv
│ ├── A01097_2024-05-16_18-53-52_FM-GenerationReport.csv
│ ├── A01097_2024-05-16_18-56-07_FM-GenerationReport.csv
│ ├── A01097_SideA_2024-05-16_15-54-16_ThermalReport.csv
│ ├── A01097_SideA_2024-05-17_07-38-52_Wash_ThermalReport.csv
│ ├── CIB Report
│ ├── ETF
│ ├── FcBirdAnalysis
│ ├── Firmware
│ ├── FlowCellFlatness
│ ├── info_00000.log
│ ├── Pressure
│ ├── UcsLogs
│ └── ZStage
├── Recipe
│ └── HVKNTDMXY.xml
├── RTA3.cfg
├── RTAComplete.txt
├── RunInfo.xml
├── RunParameters.xml
├── SequenceComplete.txt
└── Thumbnail_Images
├── L001
└── L002
barcode 정보담긴 .csv 작성
sample별로 어떤 Barcode썼는지에 대한 정보를 담고 있으면 된다.
-
Barcode Sequence를 알고 있다면,
1 2 3
[Data] Lane,Sample_ID,index,index2 1,test_sample,TGGTCCCAAG,ACGCCAGAGG
Lane확실하지 않다면 Lane column없어도 돌아감.Sample_ID정하면 그대로 fastq file name에 반영됨. -
어떤 Barcode kit 썼는지 알고 있다면,
1 2 3
[Data] Lane,Sample_ID,index 1,test_sample,SI-TT-D9
🍒Run cellranger mkfastq
1
2
3
cellranger mkfastq --id=ProjectID \
--run=/bcl/files/location/ \
--samplesheet=/location/of/barcode_info.csv
ProjectID이름으로 폴더가 새로 생기고 그 안에 결과가 저장됨.
🍒Check output fastq files
1
2
3
4
5
6
7
8
9
10
11
12
13
├── ProjectID
│ ├── ...
│ └── outs
│ ├── input_samplesheet.csv
│ ├── interop_path
│ └── fastq_path
│ ├── SampleID_BCell_S1_L001_I1_001.fastq.gz
│ ├── SampleID_BCell_S1_L001_I2_001.fastq.gz
│ ├── SampleID_BCell_S1_L001_R1_001.fastq.gz
│ ├── SampleID_BCell_S1_L001_R2_001.fastq.gz
│ ├── SampleID_TCell...
│ ├── SampleID_GEX...
│ ├── SampleID_SP...
🎀Count
demultiplexing 해서 fastq 만들었으니까 이제 GENE X CELL count matrix를 만들차례
count function을 사용하면 되는데, Sample별로 따로 돌려줘야함
🍒Prerequisite
Reference download
🔗 10X genomics download center
당영한 이야기지만 이제 read들을 reference genome 에 mappling 하고 quantification해야하니까 관련 reference들이 필요하다. 잘 다운받아서 저장해두기.
1
2
3
4
5
#GEX reference
wget "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2024-A.tar.gz"
# vdj reference
wget "https://cf.10xgenomics.com/supp/cell-vdj/refdata-cellranger-vdj-GRCh38-alts-ensembl-7.1.0.tar.gz"
feature barcode에 대한 reference도 필요한데 보통 실험하신분한테 있음. 어떤 칵테일로 바코딩했는지 정보가 필요함. 🔗10X 관련 페이지
예시로 TotalSeq-C 칵테일 썼다면 이렇게 생겼음.
1
2
3
4
5
6
7
8
9
10
11
id,name,read,pattern,sequence,feature_type
CD3,CD3_TotalSeqC,R2,5PNNNNNNNNNN(BC),CTCATTGTAACTCCT,Antibody Capture
CD19,CD19_TotalSeqC,R2,5PNNNNNNNNNN(BC),CTGGGCAATTACTCG,Antibody Capture
CD45RA,CD45RA_TotalSeqC,R2,5PNNNNNNNNNN(BC),TCAATCCTTCCGCTT,Antibody Capture
CD4,CD4_TotalSeqC,R2,5PNNNNNNNNNN(BC),TGTTCCCGCTCAACT,Antibody Capture
CD8a,CD8a_TotalSeqC,R2,5PNNNNNNNNNN(BC),GCTGCGCTTTCCATT,Antibody Capture
CD14,CD14_TotalSeqC,R2,5PNNNNNNNNNN(BC),TCTCAGACCTCCGTA,Antibody Capture
CD16,CD16_TotalSeqC,R2,5PNNNNNNNNNN(BC),AAGTTCACTCTTTGC,Antibody Capture
CD56,CD56_TotalSeqC,R2,5PNNNNNNNNNN(BC),TTCGCCGCATTGAGT,Antibody Capture
CD25,CD25_TotalSeqC,R2,5PNNNNNNNNNN(BC),TTTGTCCTGTACGCC,Antibody Capture
CD45RO,CD45RO_TotalSeqC,R2,5PNNNNNNNNNN(BC),CTCCGAATCATGTTG,Antibody Capture
Multi Config CSV 작성
Count의 input으로 사용한다. 앞서 다운받은 reference들의 위치와, 만들어둔 fastq 위치 정보 넣어서 csv 형식으로 작성하면 됨.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
[gene-expression]
reference,/source/cellranger-ref/refdata-gex-GRCh38-2020-A
[vdj]
reference,/source/cellranger-ref/refdata-cellranger-vdj-GRCh38-alts-ensembl-7.1.0
[feature]
reference,/source/cellranger-ref/CustomReferenceGenome/5gex_protein_feature_ref.csv
[libraries]
fastq_id,fastqs,lanes,feature_types
AF_GEX,/ProjectID/outs/fastq_path,1|2,Gene Expression
AF_Bcell,/ProjectID/outs/fastq_path,1|2,VDJ-B
AF_Tcell,/ProjectID/outs/fastq_path,1|2,VDJ-T
fastq_id는 위에서 fastq결과에서 _S0_L000_I1_001.fastq.gz 이 앞에 있는 부분 써주면 됨.
🍒Run cellranger count
1
2
cellranger multi --id=SampeID \
--csv=config.csv