A multi-use deep learning method for CITE-seq and single-cell RNA-seq data integration with cell surface protein prediction and imputation
| 📆 Published on | 2022 Oct 27 |
| 🎓 Authors | Justin Lakkis et al. |
| 🏫 Affiliation | University of Pennsylvania |
| 🔗 link | Nature > nature machine intelligence |
| 📂 file | paper download |
Comment / TLDR;
Research Topic
Category (General) : Method
Category (Specific) : AI, multimodal CITE-seq data
What? Paper summary (What)
- The authors developed a method named sciPENN using deep learning that can handle CITE-seq and scRNA-seq data at the same time.
- This method has three main functions, as can be inferred from the title, data integration, cell surface protein prediction, and imputation.
- In the text, the authors prepared several data for various scenarios and evaluated the performance of sciPENN compared to existing methods including Seurat and TotalVI, and claim that sciPENN’s performance is better than others.
Why? Issues addressed by the paper (Why)
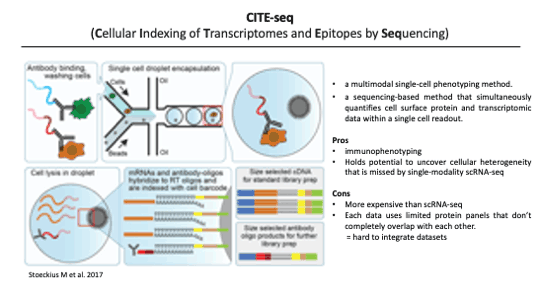 What is CITE-seq
What is CITE-seq
As an introduction, CITE-seq is an abbreviation for Cellular Indexing of Transcriptomes and Epitopes by Sequencing. It’s a sequencing method for quantifying cell surface proteins and transcriptomic data simultaneously within a single cell by using antibody-driven tags.
It is a well-known fact that proteins are much more abundant than RNA and functionally directly in cell signaling and cell-cell interactions. So Cite-seq allows us to find cellular heterogeneity that is difficult to understand with scRNA-seq alone. I think this information can better help distinguish known cell types from single-cell data, since the studies to find cell types so far have classified cell types through cell surface markers. (before bringing single-cell sequencing tech into this field)
Despite these advantages, There also exist disadvantages and technical difficulties. Producing CITE-seq data is more expensive than scRNA-seq data. The cost problem will be solved over time, but in order to solve this problem at this point, people devised some methods. The main concept of these methods is to predict the protein expression level for scRNA-seq data by understanding the relationship between RNA and protein.
Another Technical difficulty is only limited surface protein tags are created when producing the CITE-seq data, so overlapping lists can be extremely small when collecting multiple datasets. This problem must be solved for integrated analysis of multiple CITE-seq data.
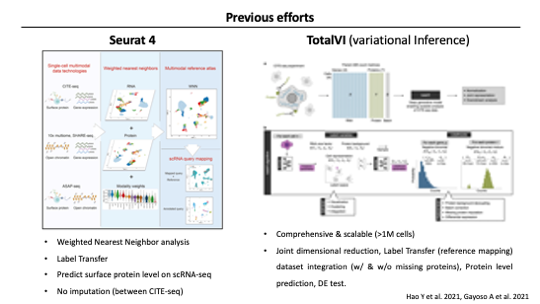
Before sciPENN, there are Seurat and TotalVI as tools to handle multimodal data like CITE-seq.
Seurat version 4 introduced Weighted Nearest Neighbor to leverage the benefits of multimodal data. WNN defines the single unified representation of single-cell multimodal data by giving weight to the modality that best describes each cell. Seurat also supports Label transfer analysis, which Annotating each query cell based on a set of reference-defined cell states. and it conducts by finding anchors between CITE-seq reference and scRNA-seq query dataset. And as One of the functions included in label transfer, it can predict surface protein levels which were inferred based on the CITE-seq reference.
Total variational inference aka TotalVI accounts for the distinct noise of each modality / and integrates multimodalities using Neural Network. It made As a comprehensive tool like suerat, so TotalVI supports many common downstream tasks including joint dimension reduction, dataset integration, Protein level prediction, and differential expression test.
As you may notice, these two preceding methods focused on utilizing multimodal data for analysis as much as possible. Because Before these methods, analysis was conducted with only one modality, and the rest of the modalities were separately reviewed even if multi-modal data was produced. Recently, the amount of published scRNA-seq and CITE-seq data is increasing rapidly. and The demand for integrated analysis with these multiple data is also increasing.
For this kind of integrated analysis, it requires highly scalable and computationally efficient methods. And it should be able to combine multiple CITE-seq data robustly and accurately even if the protein panels do not totally overlap.
How? Detailed Information (How)
Methodology
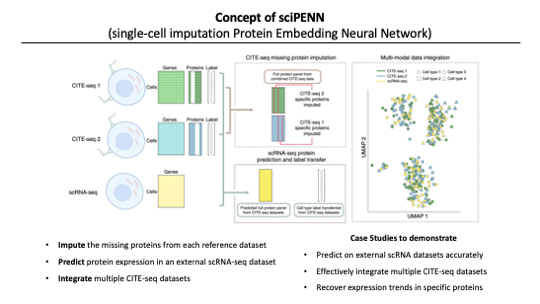
So.. Today’s authors developed sciPENN.
It’s a deep learning framework. It learns from one or more CITE-seq reference datasets for predicting and imputing protein expression, integrating datasets in a low-dimension embedding and merging multiple CITE-seq data even when their protein panels do not totally overlap. it’s much more accurate, robust, and faster than previous methods, according to this paper.
After developing the methods, the authors want to test the performance of this method. So they made various scenarios of datasets to demonstrate the better performance of sciPENN compared with TotalVI and Seurat4.
Results
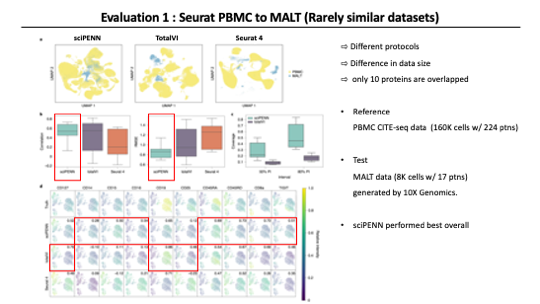
For the first evaluation, the authors used two data sets to demonstrate that sciPENN performed well even when single-cell protocols differ substantially between the datasets.
As a reference, they used the Human peripheral blood mononuclear cells (PBMC) CITE-seq data reported in Seurat 4 paper. It includes 224 proteins. For the test set, They used Mucosa-Associated Lymphiod Tissue dataset, which includes 17 proteins. And it’s generated by 10X genomics. Among the 17 proteins in the MALT datasets, 10 overlapped proteins in PBMC data.
The authors evaluated the performance of the methods through the following approaches. First, they embedded reference and query data together into a single latent space using each method. And evaluated how well they integrate the two datasets. Second, they examined the accuracy of protein expression prediction in each method. All three methods support the function of predicting protein levels in query data based on reference data. They quantified the prediction accuracy by Pearson correlation and Root Mean Squared Error values. Lastly, They examined feature plots for individual proteins predicted by each method. And calculated the correlation between the gold standard and predicted protein expression.
As a result in this case, Owing to the substantial differences between the PBMC and MALT data, all methods struggled to fully mix the two datasets together. sciPENN did the best at integrating two datasets as you can see in the UMAP. And sciPENN achieved the highest protein prediction accuracy among all proteins as quantified by both correlation and RSMEs. For the feature plot correlations, sciPENN performs the best overall. But totalVI performed well in some proteins.
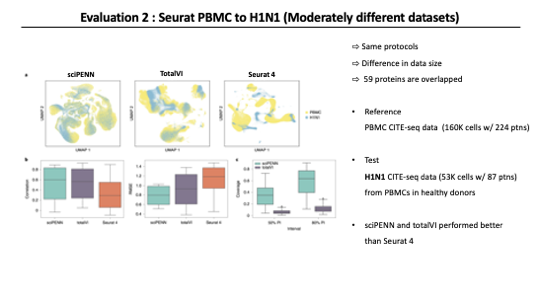
As a second evaluation, they considered a situation where the query set is Moderately different from the reference set. So they reused Seurat 4 PBMC dataset as the reference and used H1N1 influenza dataset as the query. H1N1 dataset is CITE-seq data that includes 87 proteins from PBMCs in healthy donors.
In this modertate sinario, sciPENN and TotalVI show reasonably well-mixed data in embedding space. But Seurat 4 still can not mix batches of these datasets. It was not effective. sciPENN and totalVI predicted protein expression more effectively than Seurat4.
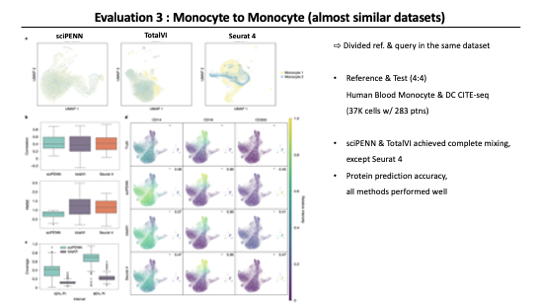
Move on to the next evaluation, This time Authors considered a more Even-handed balance between query and reference sets. So they generated the Human Blood monocyte and Dendritic cell CITE-seq dataset from 8 samples. and allocated 4 samples to the reference and the other 4 samples to the query.
As a result of this scenario, sciPENN achieved complete mixing of two datasets and toatlVI also achieved nearly complete mixing, but Seurat4 did not mix datasets as well as the other methods. And then for the protein prediction accuracy, all three methods performed highly effectively in this analysis. Lastly, they checked the feature plot for monocytes and DC markers and All three methods performed well.
At this point, I doubted that they used only single-modality without using WNN when integrating using seurat 4. So I searched their code and confirmed they used WNN.
So.. Through all these evaluations, it was confirmed that the performance of sciPENN is better than Seurat in integrated analysis so far.
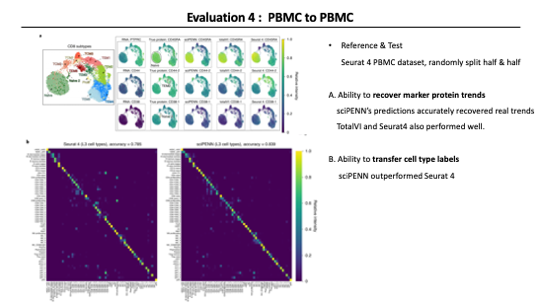
To evaluate other aspects of sciPENN, in this case, The Authors randomly split the PBMC data into reference half and query half.
First, they focused on the CD8T population to evaluate the recovering ability of marker protein trends. They selected three “features” that divide the subtypes of CD8T, but not by RNA gene expression alone. In other words, they selected “features” that can distinguish subtypes by protein expression pattern. These feature plots show that sciPENN’s protein predictions accurately recovered real trends and TotalVI and Seurat4 also performed well.
Next, they evaluated sciPENN’s and Seurat 4’s ability to transfer cell type labels from CITE-seq reference to scRNA-seq query. Figure B shows the cell type prediction accuracy of each method. Overall, sciPENN outperformed Seurat 4 for predicting cell type labels, despite using the labels originally assigned by Seurat 4.
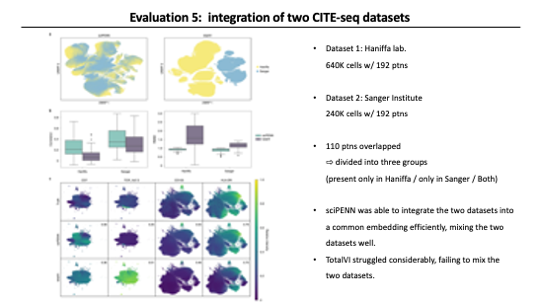
In the last evaluation, They prepared for a task combining multiple CITE-seq datasets.
Different CITE-seq datasets can have different protein panels, so the key is how well the method fills in missing proteins in each dataset. Seurat was not included in this evaluation because it is not able to impute protein expression level from RNA expression. (It only able to map a reference CITE-seq dataset to a query RNA dataset)
For this evaluation, They used Two CITE-seq datasets. Both datasets were generated from healthy donors and patients infected with COVID-19, but generated at different institutions. One was made at Haniffa Lab and the other at Sanger Institute. 110 proteins overlapped between these two datasets. To make the situation of merging two CITE-seq datasets with partially overlapping protein panels, They randomly partitioned these 110 proteins into three groups: proteins present only in Haniffa, only in Sanger, and present in both.
And then, They trained both sciPENN and totalVI with these datasets and impute the missing protein expression for each dataset. As a result, Looking at figure A, sciPENN did well mixing two datasets, but totalVI failed to mix.
After that, They examined protein imputation accurary. sciPENN clearly outperformed totalVI in both measurements.
Lastly, They examined the feature plots for few proteins. First two proteins are Sanger Only proteins imputed into Haniffa dataset. The other two proteins are Haniffa-only proteins imputed into the Sanger dataset. As you can see both methods struggled and totalVI failed to predict the proteins in Haniffa datset. They said it might be caused by the sequencing depth. The Sequencing depth in Haniffa data is only up to 50% of Sanger data. In the latter two proteins, both methods did well.
And? Conclusions
The author’s conclusions
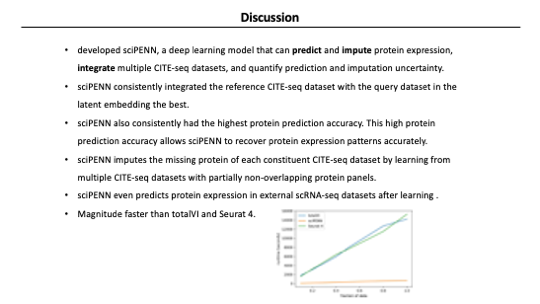
In summary, They developed sciPENN to address the Challenges of CITE-seq data analysis using multiple datasets. It is a deep learning model that can predict and impute protein expression and integrate multiple CITE-seq datasets.
They showed us that sciPENN consistently performed best in integration and protein level prediction across different evaluations.
sciPENN also can impute the missing protein in each CITE-seq dataset with different protein panels. TotalVI also has this feature, but sciPENN showed better performance.
The novel feature is that sciPENN can predict protein in external scRNA-seq datasets after learning from multiple CITE-seq reference datasets. Since this feature is not supported by other methods, it was not evaluated in this paper. I think it’s a pretty useful function. We can convert scRNA-seq data to CITE-seq data If this works well.
And the last advantage of sciPENN is it’s faster than any other method. The figure on the right is a graph measuring how long the process takes for each method while increasing the data size. Seurat and totalVI showed a linear increase in runtime as the data size increased, but sciPENN showed a much lower slope.
My Conclusion
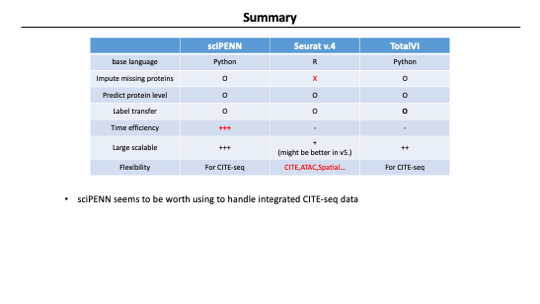
This new CITE-seq integration tool sciPENN is interesting because there was a function to impute the missing protein level that the Seurat doesn’t support. I also like its good performance in integrating query and reference datasets into an embedding space.
I already knew that Seurat’s batch correction performance was not the best through several benchmarking papers. But the results of integrating multimodal data in this paper were worse than I know.
One thing that I think could be a better evaluation is checking whether it is clustered properly to divide the cell type. I wonder if sciPENN actually can help find cell types by highlighting the characteristics of RNA and protein, beyond the batch correction.
Anyway. I’ve mainly used seurat in my analysis so far, but sciPENN seems to be worth using to handle integrated CITE-seq data. I would try to apply it to my research.